



INTRODUCCIÓN
Las diferentes opciones de tratamiento de la disfunción eréctil se han modificado de forma significativa durante los ultimos 30 años, y se ha cambiado del tratamiento psicosexual y las prótesis de pene (1970), pasando por la revascularización, los mecanismos de vacío y las inyecciones intracavernosas (1980), al desarrollo de tratamientos transuretrales y orales (1990). La introducción del primer inhibidor de la fosfodiesterasa tipo 5 (PDE5), el sildenafilo, en 1998, ha revolucionado el tratamiento de los varones con disfunción eréctil de diferentes etiologías. La segunda generación de inhibidores de la PDE5, vardenafilo y tadalafilo, y el desarrollo de nuevos inhibidores de la PDE5 hace que este campo esté en continua actualización.
El objetivo de la presente revisión es comparar, desde el punto de vista farmacológico, los 3 inhibidores y establecer sus diferencias farmacológicas, para una mejor y más racional utilización de éstos en la práctica clínica diaria1.
EVALUACIÓN COMPARATIVA DE LOS INHIBIDORES DE LA PDE5
Aunque los 3 inhibidores de la PDE5 actualmente disponibles, sildenafilo, vardenafilo y tadalafilo, han demostrado ser efectivos en el tratamiento de la disfunción eréctil, hay una serie de diferencias entre los compuestos en lo que se refiere a su selectividad y su especificidad por la inhibición de la PDE, con consecuencias especialmente en el perfil de seguridad pero también en las disparidades biofarmacéuticas y farmacocinéticas que afectan de forma importante al perfil de eficacia de estos fármacos.
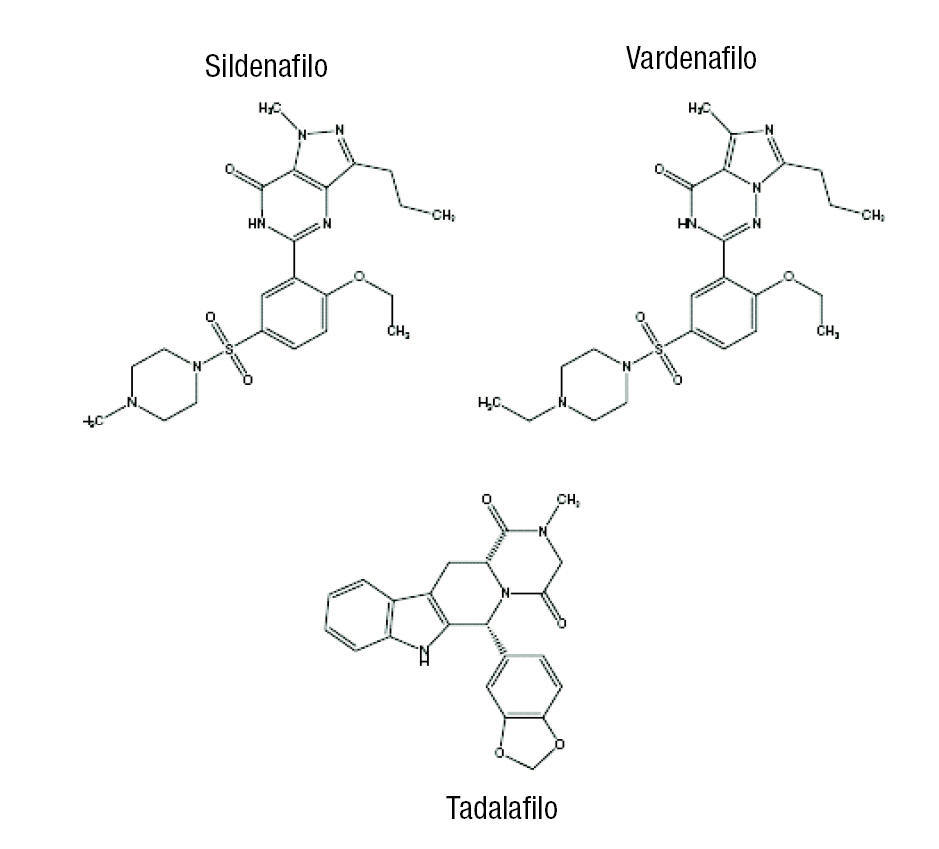
El sildenafilo y el vardenafilo son muy similares en términos de estructura química, mientras que el tadalafilo, con una estructura de metidiona, difiere de forma marcada del sildenafilo y el vardenafilo (fig. 1).
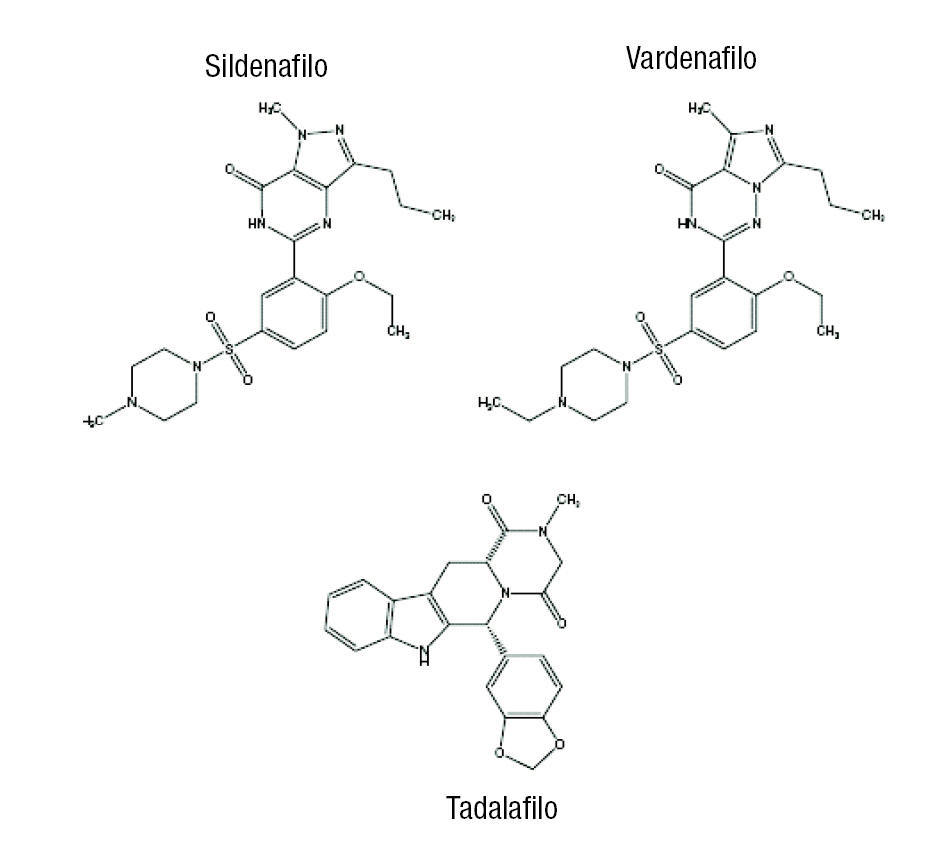
Figura 1.
Estas similitudes y diferencias químicas también se reflejan en las similitudes y las diferencias de su farmacocinética clínica (tabla 1).

Según el sistema de clasificación biofarmacéutica de la Food and Drug Administration (FDA), el sildenafilo es un fármaco de clase 1, con una solubilidad y una permeabilidad altas, mientras que el vardenafilo y el tadalafilo son fármacos de clase 2, con una baja solubilidad y una alta permeabilidad.
El sildenafilo y el vardenafilo tienen solamente una limitada biodisponibilidad oral, principalmente debido a un metabolismo presistémico extenso en la pared digestiva y al metabolismo hepático de primer paso vía CYP3A4 y/o CYP3A5.
La menor y variable biodisponibilidad oral del vardenafilo, comparada con el sildenafilo, parece ser el principal factor en su mayor variabilidad interindividual en la aclaración oral y la exposición sistémica, en comparación con el sildenafilo y el tadalafilo.
No se ha descrito la biodisponibilidad oral del tadalafilo, pero el 36% de la dosis oral parece ser absorbida a traves del tracto gastrointestinal, de forma similar al sildenafilo y el vardenafilo; sin embargo, CPY3A es también la mayor enzima metabolizante para el tadalafilo, lo que sugiere que también está sujeto a metabolismo presistémico.
Los 3 inhibidores de la PDE5 se absorben rápidamente tras su administración oral, con concentraciones pico que se consiguen ligeramente más rápido con el vardenafilo frente al sildenafilo y al vardenafilo (tabla 1).
Aunque no se ha establecido una clara relación concentración-efecto para cualquiera de los 3 inhibidores de la PDE5, la rápida absorción se considera un requisito para una rápida aparición de su eficacia.
La administración de una comida con alto contenido en grasa no tiene efecto en la tasa y la extensión de la absorción del tadalafilo, pero disminuyó la tasa de absorción del sildenafilo y el vardenafilo. Aunque esto se ha interpretado como una ventaja para el tadalafilo, ya que elimina la necesidad de coordinar el tiempo de las comidas alrededor de la actividad sexual, está poco claro si el incremento en el tiempo hasta la máxima concentración plasmática (Tmáx) inducido por el alimento y la disminución de la concentración máxima (Cmáx) del sildenafilo y el vardenafilo tienen relevancia clínica.
Los 3 fármacos son lipofílicos y tienen un volumen de distribución mayor que el volumen de agua corporal total, lo que indica captación tisular y unión. Es más, los 3 fármacos se unen altamente a proteínas, con concentraciones de fracciones plasmáticas libres de sólo un 4 a un 6%. La principal vía de eliminación de todos los inhibidores de la PDE5 es el metabolismo hepático, con una eliminación renal de fármaco no alterado del 1% o menos de las vías de eliminación. Además de CPYA3, como principal enzima de metabolización del fármaco para los 3 inhibidores, CYP2C9, 2C19 y 2D6 tambien contribuyen al metabolismo del sildenafilo y 2C9 contribuye al metabolismo del vardenafilo. Tanto el sildenafilo como el vardenafilo tienen metabolitos activos que alcanzan concentraciones plasmáticas lo suficientemente altas como para contribuir al perfil de eficacia global y de seguridad de estos fármacos.
Basado en su relativamente alta aclaración sistémica tras su administración sistémica, el sildenafilo y el vardenafilo se pueden clasificar como fármacos aclarados de forma no restrictiva, con extracción hepática intermedia y alta2.
De forma consistente con esta clasificación biofarmacéutica, tanto el sildenafilo como el vardenafilo tienen una biodisponibilidad oral relativamente baja, debido a la inactivación extensa del primer paso.
Por el contrario, el tadalafilo puede clasificarse como un fármaco con una tasa de extracción hepática baja, por lo que probablemente se aclara de forma restrictiva. Los volúmenes de distribución relativamente comparables, junto con las diferencias sustanciales en elaclaramiento sistémico entre los inhibidores de la PDE5, ocasionan las diferencias en la vida media de eliminación: de 3 a 5 h en el sildenafilo y el vardenafilo en comparación con las 17,5 h del tadalafilo.
La vida media del tadalafilo ocasiona un período de respuesta largo en comparación con el sildenafilo y el vardenafilo. Esta mayor ventana terapéutica del tadalafilo requiere menor tiempo para la efectividad, lo que se ha interpretado como una ventaja para tener la opción de una actividad sexual más espontánea. El tadalafilo, sin embargo, se ha detectado en plasma incluso 5 días después de su administración oral, debido a su larga vida media. Esto sugiere la posibilidad de acumulación si se toma regularmente y en intervalos cortos, lo que puede suponer un riesgo incrementado de efectos adversos si se usa excesivamente.
Durante el uso clínico normal, ninguno de los 3 inhibidores de la PDE5 se ha idenfificado para inhibir el metabolismo del fármaco inducido por la isoenzima CYP, incluyendo CPY3A o CYP2C. Dado a que el metabolismo vía CYP3A es la mayor vía de eliminación de los 3 fármacos, todos los inductores e inhibidores de la actividad de CYP3A tienen el potencial de interferir con la eliminación del sildenafilo, el vardenafilo y el tadalafilo.
Esta potencial interacción se ha verificado clínicamente en inductores de la actividad de CYP3A sólo para la rifampicina y el tadalafilo. Para los inhibidores potentes del metabolismo mediado por CYP3A, se observó que la exposición sistémica estaba aumentada en el ritonavir, el indinavir, el saquinaqvir, la eritromicina y el ketoconazol.
El mosto, como un selectivo inhibidor de metabolismo de CYP3A, mediado en el tubo digestivo, también aumentó la exposición sistémica al sildenafilo y el vardenafilo. De forma similar, la exposición sistémica a ambos fámacos estaba aumentada durante el tratamiento concomitante con la cimetidina, un inhibidor no específico de CYP3A.
De forma notable, el ritonavir, un inhibidor de CYP3A, así como del metabolismo mediado por CYP2C, tiene un efecto inusualmente alto en el vardenafilo: aumenta la exposición sistémica una media de 49 veces con valores individuales de hasta 300 veces, muy probablemente como consecuencia de una inhibición simultánea de CYP3A y CYP2C9 como pricipales vías de eliminación de vardenafilo.
El efecto del ritonavir en el sildenafilo fue mucho menos pronunciado (11 veces), dado que otras vías de eliminación compensatorias, junto con CYP3A y CYP2C9, estaban todavía disponibles. Para CYP3A, pero no para CYP22, el incremento en la exposición sistémica fue sólo de 2,6 veces, en el caso del tadalafilo. Sin embargo, a diferencia de la información de la Unión Europea, en Estados Unidos sólo se recomienda una reducción del 50% de la dosis, pero no existe contraindicación para el uso del vardenafilo y el ritonavir.
Como fármaco clase 2, la tasa de absorción del tadalafilo disminuyó hasta en un 30% cuando se coadministró con antiácidos. No se observó ningún efecto con el vardenafilo y el sildenafilo, que se incluyen como fármacos de clase 1, que tienen sustancialmente una mayor solubilidad que el tadalafilo (0,11 mg/ml frente a prácticamente insolubles en agua)3,4.
La actividad CYP3A y CYP2C parece ser dependiente de la edad, según estudios in vitro con cromosonmas hepáticos de rata y humanos, con una reducción de la actividad en individuos mayores en comparación con los jóvenes5,6. Esta disminución de la actividad metabólica se refleja en un incremento correspondiente de la exposición sistémica de los 3 inhibidores de la PDE5; se requiere una reducción de la dosis para el sildenafilo y el vardenafilo en pacientes mayores.
De forma similar, se pueden esperar diferencias dependientes de la raza en la farmacocinética de los 3 inhibidores de la PDE5, basándose en las diferencias raciales conocidas de la actividad de CYP3A4/57,8. Hasta la fecha, el único efecto relacionado con la raza de los inhibidores de la PDE5 es un incremento de la exposición sistémica en mexicanos, secundaria a una conocida disminución de la actividad de CYP3A9.
No se han descrito diferencias en función del sexo en la farmacocinética en ninguno de los 3 inhibidores de la PDE5, lo que parece estar de acuerdo con la literatura médica10,11.
La disminución grave de la función renal resulta en un incremento de la exposición sistémica en los 3 fármacos, lo cual precisa una disminución de la dosis para el sildenafilo y el tadalafilo en los pacientes afectados.
Dado que la eliminación renal contribuye sólo en un 1% o menos a la aclaración sistémica para todos estos fármacos, es probable que el aumento de la exposición sistémica sea la consecuencia de una inhibición directa de la función hepática, secundaria a afección renal, probablemente mediante la acumulación de sustancias endógenas que inhiben las enzimas hepáticas metabolizantes del fármaco y/o los cambios en la vía hemodinámica que afectan al flujo sanguíneo del hígado.
La afectación hepática leve o moderada aumenta la Cmáx y el área bajo la curva (AUC) en el sildenafilo y el vardenafilo, pero no tiene efecto significativo en la farmacocinética del tadalafilo. Hasta la fecha, la insuficiencia hepática grave no se ha estudiado en relación con ninguno de los 3 inhibidores de la PDE5.
Como se ha señalado anteriormente, los inhibidores orales actualmente disponibles se pueden diferenciar sobre la base de sus perfiles biofarmacéuticos y farmacocinéticos.
La respuesta a los inhibidores de la PDE5, sin embargo, no sólo está determinada por su selectividad, especificidad y potencia relativa, así como por su perfil farmacocinético, sino que también parece estar afectada por factores genéticos.
Recientes investigaciones sugieren que pacientes homocigóticos para una variente de alelos de un polimorfismo de un único nucleótido en el gen que codifica la subunidad 3 de la proteína G (GNB3) tienen una odds ratio de 10 de presentar una respuesta eréctil positiva tras la administración de sildenafilo, en comparación con los pacientes que portan al menos un alelo tipo Wild 1. Esto se ha traducido en una respuesta positiva del 90,9% en los individuos homocigóticos para la variante del alelo, frente a una respuesta del 50% para los portadores del alelo Wild tipo 112.
Los inhibidores actualmente disponibles de primera y segunda generación (sildenafilo, vardenafilo y tadalafilo) han emergido como tratamiento de primera línea para la disfunción eréctil, por su seguridad, eficacia clínica y mejoría importante de la calidad de vida en varones con disfunción eréctil de diferentes etiologías.
Son necesarios más estudios acerca de su farmacología clínica, así como de su eficacia y seguridad, para complementar las bases científicas del uso racional, la prescripción basada en la evidencia y las decisiones de la dosificación, particularmente en las situaciones en las que se produce un marketing a gran escala para el uso directo al consumidor de estos productos.
Correspondencia:
Dr. A. Astobieta Odriozola.
Servicio de Urología Cínica. Clínica Virgen Blanca.
Maestro Mendiri, 2. 48009 Bilbao. Vizcaya. España.
Correo electrónico: anderastobieta@terra.es