




La supervivencia media de los pacientes de Fibrosis Quística (FQ) ha aumentado considerablemente en la última década. Esto gracias a la intervención precoz y agresiva de las alteraciones respiratorias y de la nutrición; y al manejo de los pacientes en centros de FQ con atención multidisciplinara y basados en protocolos escritos y sistematizados. El conocimiento del defecto básico de la enfermedad, la falla cuantitativa o funcional de la proteína CFTR, de la que depende el equilibrio hidroelectrolítico en el lumen de las vías respiratorias, y de otros epitelios, ha llevado al desarrollo de terapias dirigidas a su corrección, que se encuentran en etapas avanzadas de investigación. Se ha abierto así, la esperanza de contar con terapias dirigidas a la causa de la FQ y sólo a aliviar sus consecuencias.
The median survival of Cystic Fibrosis patients has increased remarkably in the last decade, due to precocious and aggressive respiratory and nutritional intervention, and the multidisciplinary management of the disease based on systematized protocols. The knowledge of the basic defect of the disease, the cuantitative or functional fault of the CFTR protein, on which the hidroelectrolytic balance of the epitelial airways depends, has allowed the development of specific therapies targeted to its correction, in advanced stages of investigation. The hope has been opened to have therapies directed to the cause of the Cystic Fibrosis and not only to alleviate its consequences.
La Fibrosis Quística (FQ) es la enfermedad genética letal más frecuente en la población blanca, aunque se describe en todas las etnias, con una frecuencia que varía de 1:2000 recién nacidos en caucásicos, hasta 1:31000 recién nacidos en asiáticos (1). Continúa siendo causa de importante daño en morbilidad biológica y psicológica para el paciente y su familia, y un serio desafío para los sistemas de salud.
Este artículo tiene como objetivo actualizar la información de la literatura en los aspectos etiopatogénicos, diagnósticos y terapéuticos de esta enfermedad.
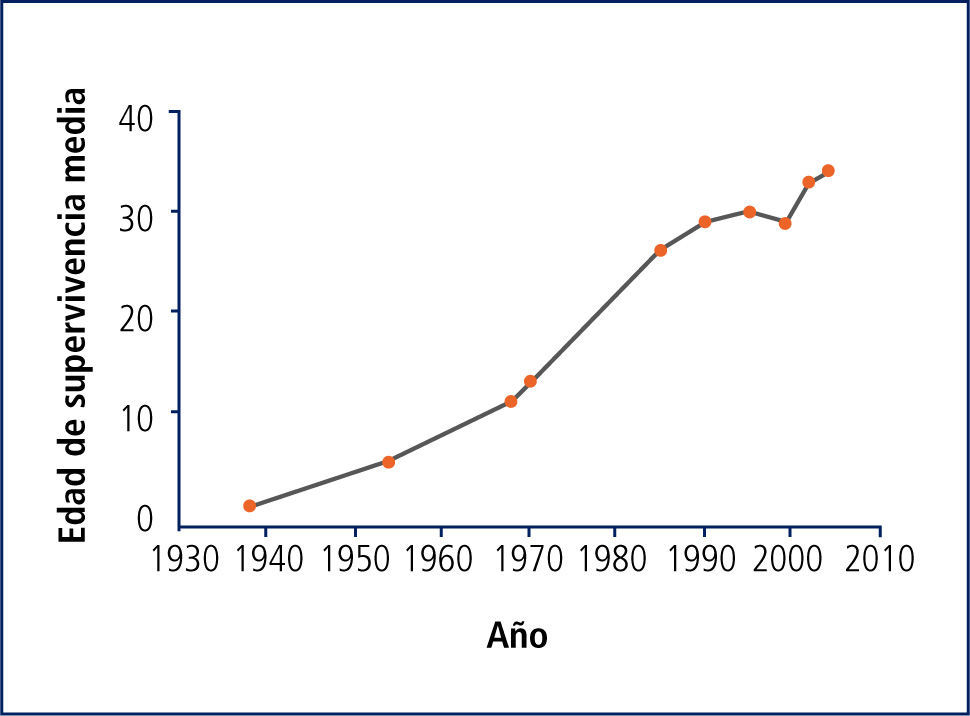
Sobrevida de la fq en la actualidadDesde su descripción en 1938, las curvas de supervivencia media han mejorado notablemente en los países desarrollados, llegando a los 37 años en EE.UU. en 2008, y a 50 años en Dinamarca (2) (Figura 1).

Las razones de este aumento son variadas, las principales son la intervención precoz y agresiva de las complicaciones respiratorias, el manejo nutricional riguroso evitando la malnutrición y fundamentalmente el desarrollo de Centros de FQ con enfoque multidisciplinario, donde el paciente recibe atención integrada de todos los aspectos de su patología, con tratamientos basados en la evidencia, sistematizados y protocolizados en algoritmos de tratamiento.
En Chile no hay datos de sobrevida media, sin embargo, desde el inicio del Programa Nacional de FQ en 2002, la proporción de pacientes mayores de 15 años aumentó de 26% en 2003 a 40% en 2008, y los mayores de 18 años aumentaron de 7,7% en 2003 a 23% en 2008, reflejando una mejoría importante en su pronóstico (3).
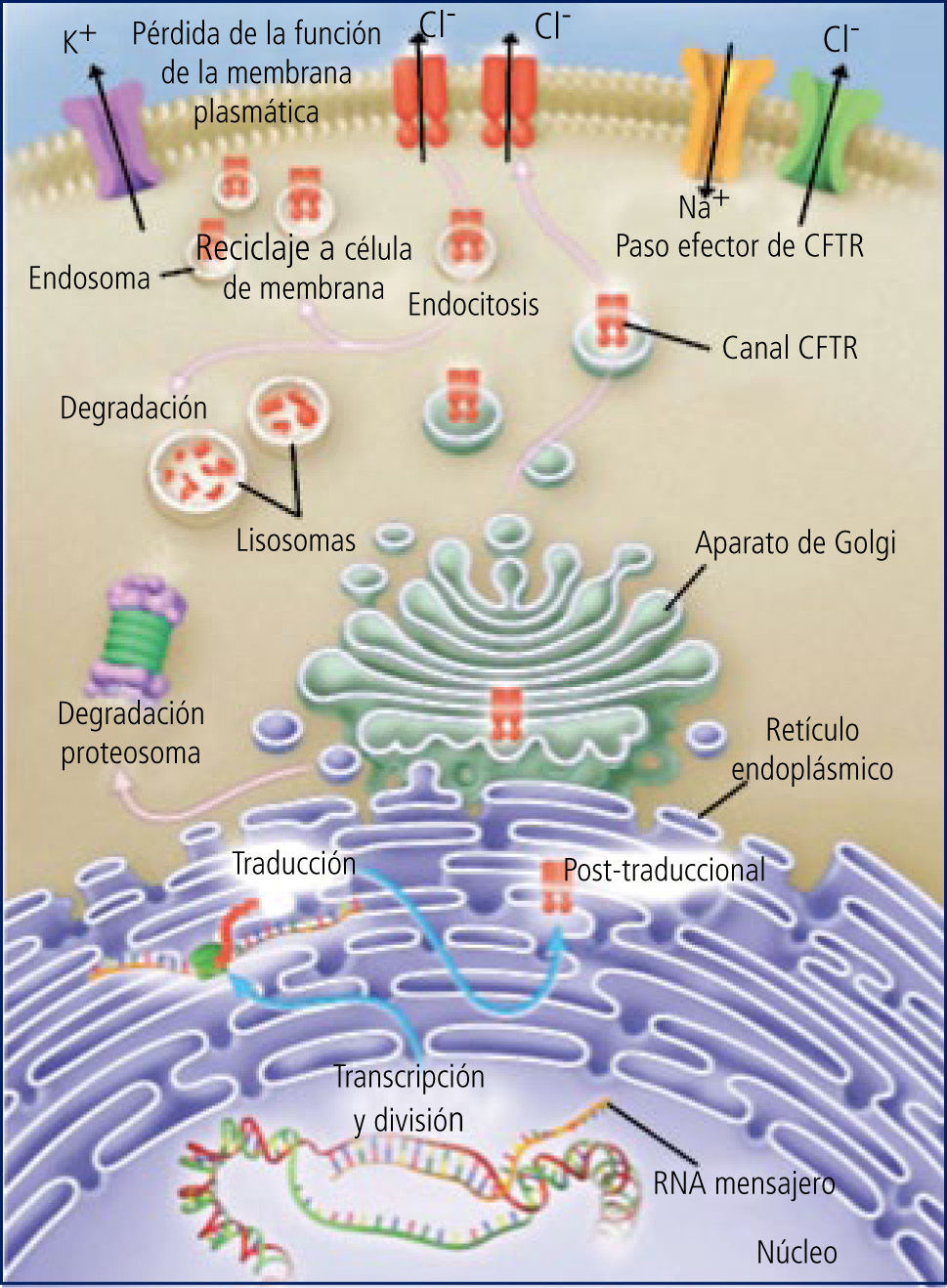
GenéticaDesde el descubrimiento del gen de FQ en el brazo largo del cromosoma 7 en 1989, se han descrito más de 1800 mutaciones, de las cuales la mayoría son variantes que no producen enfermedad FQ. Alrededor de 30 a 40 de ellas inducen la falta de producción, o la producción defectuosa de la proteína CFTR (del inglés cystic fibrosis transmembrane regulator), que regula el paso del ión cloro en las membranas celulares, por lo que también se le conoce como el canal del cloro (Figura 2).

El defecto se hereda de manera autosómica recesiva, por lo que se requiere que ambos padres sean portadores del gen defectuoso y la probabilidad de tener un hijo con FQ es del 25% en cada embarazo.
La mutación más frecuente entre individuos blancos del hemisferio norte es la pF508del, presente en el 60 a 70% de los casos, y que produce la pérdida de fenilalanina en la posición 508 de la proteína. En Europa su frecuencia varía desde el 22% en judíos Ashkenazíes hasta el 90% en Dinamarca.
El CFTR se encuentra en la mayoría de los epitelios, lo que explica que la FQ sea una enfermedad multisistémica, con compromiso variable de vías aéreas y parénquima pulmonar, conductos pancreáticos, intestino, canalículos excretores de las glándulas sudoríparas, conductos biliares y conductos deferentes.
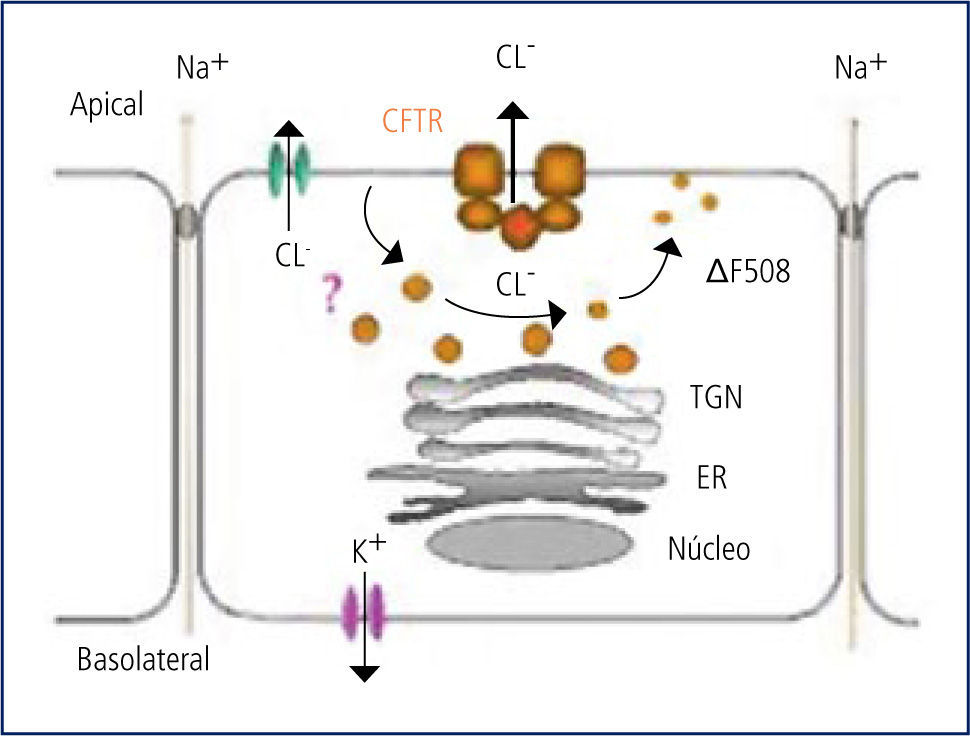
El CFTR es una proteína de alto peso molecular, que funciona como un canal para el paso del ión cloro, dependiente de ATP. De su correcto funcionamiento depende la hidratación del lumen correspondiente. En la vía aérea, la hidratación del moco luminal depende del equilibrio entre las fuerzas que desplazan sodio (y por ende agua) desde el lumen hacia el intersticio (bomba Na/K en la membrana basal) y el cloro que entra a la célula por transporte activo desde el intersticio junto con Na y K y que “escapa” hacia el lumen a través del canal del cloro que se abre por mediación del cAMP en la superficie luminal de la célula. Este escape del cloro arrastra sodio por diferencia de carga eléctrica, el que mantiene la hidratación del lumen. Este líquido de la superficie epitelial permite la correcta disposición y funcionamiento de los cilios. (Figura 3).

En la vía aérea de los niños con FQ, la falla del canal del cloro lleva a que éste se acumule en el intracelular, produciéndose un desequilibrio con reabsorción marcada del sodio intraluminal, el que arrastra al agua. Esto lleva a una disminución de la altura del líquido en la superficie epitelial, con aplastamiento y mal funcionamiento de los cilios y el consecuente daño al transporte mucociliar.
En Chile se han encontrado 14 mutaciones, con una tasa de detección del 42%, entre las cuales la mas frecuente es la pF508del, encontrada con una prevalencia de 30%. Otras 4 mutaciones tienen prevalencia sobre 1 %, y son p.R334W, p.G542X, c.3849+10Kb C>T, y p.R553X (3).
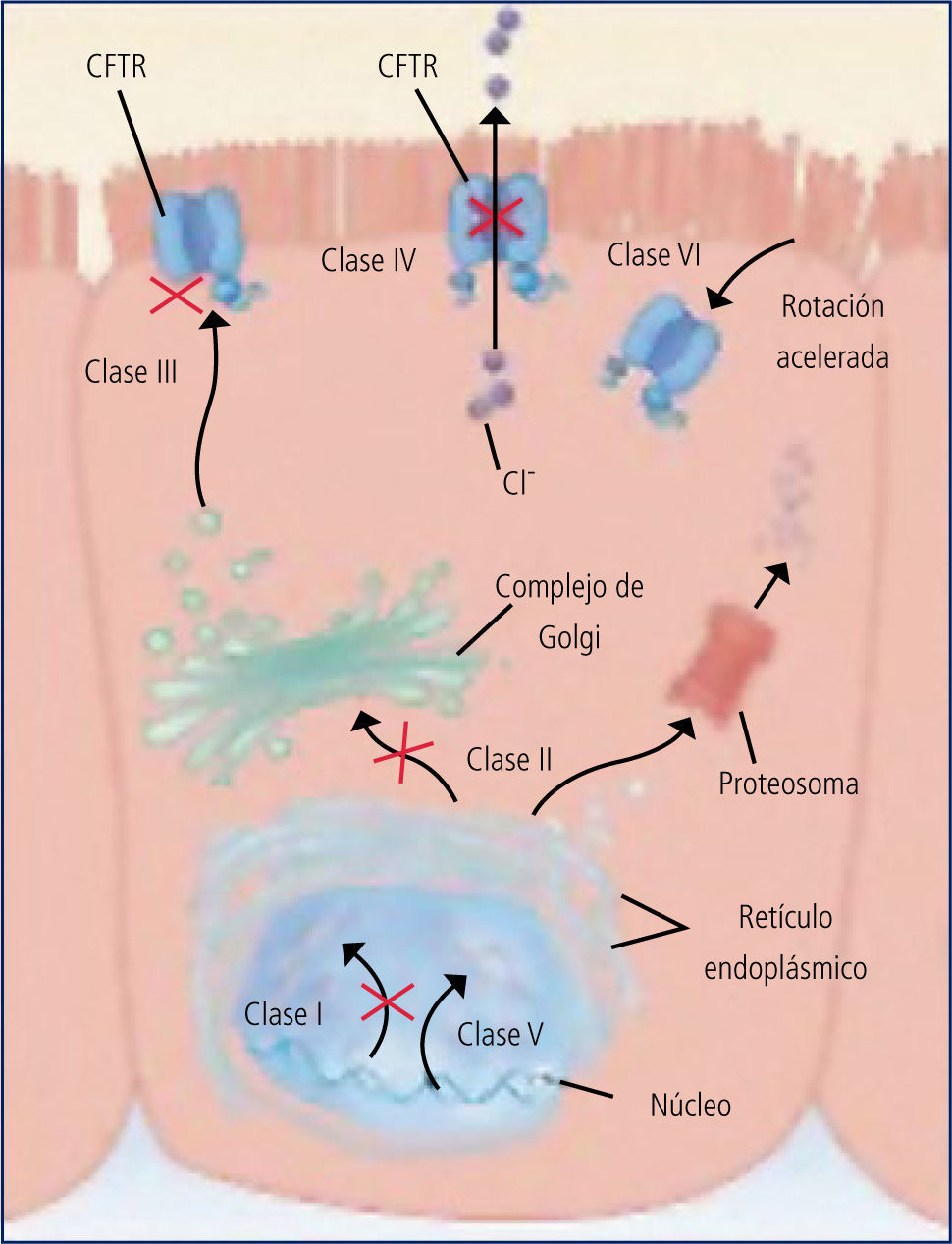
Las distintas mutaciones originan fallas en distintos niveles de la producción del CFTR y se clasifican en 6 clases: (Figura 4).
- *
Clase 1 : defecto de síntesis (G542X).
- *
Clase 2: defecto de maduración al pasar al retículo endoplásmico (pF-508del, N1303K).
- *
Clase 3: bloqueo de activación (G551D, G551S).
- *
Clase 4: defecto de la conducción (R117H, R334W).
- *
Clase 5: empalme incorrecto (3849+10Kb C T).
- *
Clase 6: defecto de regulación (G551 D).

Las mutaciones de clase 1, 2 y 3 se asocian a fallas severas de producción del CFTR y por lo tanto a fenotipos más graves, en los que a la patología respiratoria se asocia la insuficiencia pancreática con esteatorrea y malabsorción. La presencia de una mutación de las clases 4, B o 6 en uno de los alelos, se asocia a fenotipos leves, sin falla pancreática y de mejor pronóstico.
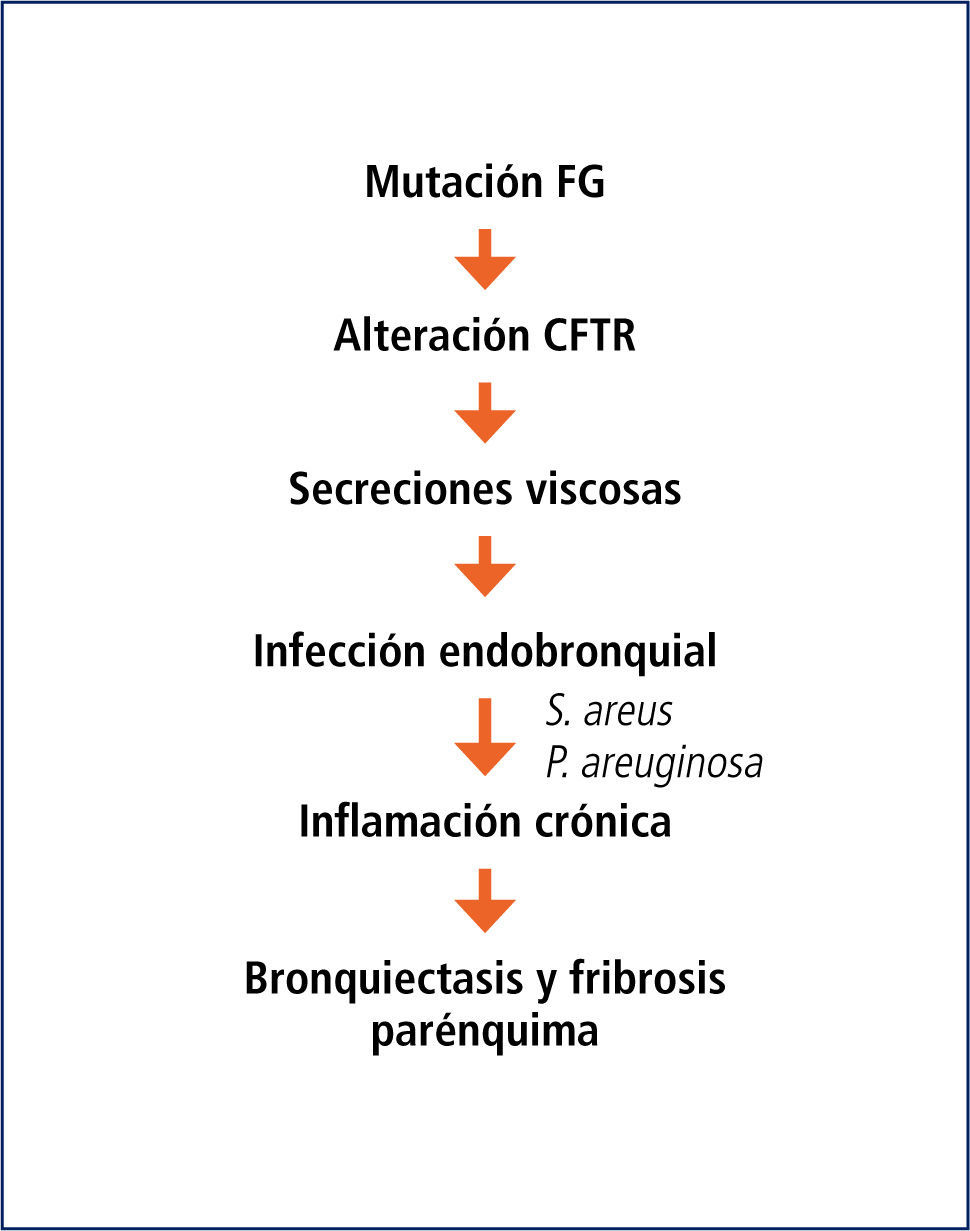
Etiopatogenia del daño pulmonarSi bien es una enfermedad multisistémica, la letalidad depende del compromiso respiratorio.
En la vía aérea de los niños con FQ, la falla del CFTR lleva a que el ión cloro se acumule en el intracelular, produciéndose un desequilibrio con reabsorción marcada del sodio intraluminal, el que arrastra agua desde el lumen hacia el intersticio del epitelio respiratorio. Esto lleva a una disminución de la altura de la capa líquida en la superficie epitelial, con deshidratación del contenido luminal y aplastamiento y mal funcionamiento de los cilios y el consecuente daño al transporte mucociliar. La consecuencia es la producción de un mucus deshidratado, extremadamente viscoso y espeso, que se adhiere a los bronquíolos y bronquios, obstruyéndolos progresivamente.
Esta secreción es especialmente susceptible a la colonización con bacterias, inicialmente Haemophylus influenzae y Staphylococcus aureus y luego Pseudomonas aeruginosa. La infección bacteriana endobronquial se hace crónica, especialmente por S. aureus y P. aeruginosa, generando una respuesta inflamatoria persistente y muy intensa, con llegada de gran cantidad de neutrófilos, los que contribuyen al daño de la pared bronquial y el parénquima pulmonar y, liberando sus enzimas proteo-líticas (elastasa) y factores oxidantes. La acumulación de ADN de los neutrófilos y las bacterias en el lumen le dan la gran viscosidad a las secreciones, las que se transforman en un verdadero “pegamento”. Con el paso del tiempo, el daño pulmonar se manifiesta en la formación de grandes impactos mucosos, bronquiectasias y fibrosis (Figura 5).

La infección endobronquial por Pseudomonas aeruginosa es el principal factor que marca el pronóstico de la enfermedad. Inicialmente la infección es por bacterias planctónicas, susceptibles de ser erradicadas de la vía aérea con tratamiento antibiótico agresivo, y por lo tanto se presenta intermitentemente en la evolución de los cultivos de secreción bronquial del paciente. Si no se detecta y se trata adecuadamente, el número de bacterias aumenta hasta un punto crítico en el que se gatilla un cambio fenotípico, que las hace secretar un biofilm de alginato, el que perpetúa la infección, al dotarlas de un verdadero escudo que las protege del ataque de anticuerpos, neutrófilos y antibióticos. Este fenómeno de cambio del fenotipo de P. aeruginosa se ha llamado microevolución y es el principal responsable del rápido deterioro clínico, de la caída de la función pulmonar y la muerte de los enfermos. Se ha estimado que el tiempo necesario para que la infección se haga inerradicable sería aproximadamente de 1 año (4).
Precocidad del daño pulmonarSe sabe que los pulmones del paciente FQ son completamente normales al nacer, y que al momento del diagnóstico la función pulmonar ya está deteriorada. Este deterioro progresa en el tiempo, a pesar de la terapia agresiva. Existían dudas en cuanto a la precocidad con la que se produce el daño después del nacimiento. El trabajo del Dr. P. Sly en Australia, en 57 lactantes asintomáticos diagnosticados con pesquisa neonatal, muestran daño pulmonar ya a los 3 meses de vida, evidenciado cambios en la tomografía pulmonar de alta resolución en el 80% de los casos (bronquiectasias en 18%, engrosamiento de pared bronquial en el 45%, atrapamiento aéreo en el 67%), infección bacteriana por S.aureus y P. aeruginosa en 20%, y evidencias de inflamación pulmonar en el lavado bronquioalveolar con niveles altos de IL-8 en el 77% de los casos, y aumento de actividad de elastasa de neutrófilos en el 30% (5).
De éste y otros estudios previos, se demuestra que el fenómeno inflamatorio pulmonar es parte de la enfermedad, muy precoz y en gran parte independiente de la presencia de infección bacteriana endobronquial. De aquí la importancia del diagnóstico lo más temprano posible. Lamentablemente, si el diagnóstico se basa en criterios de sospecha clínica, es muy raro que éste se logre hacer antes de los 6 meses de edad.
Avances en el diagnóstico de la fqEs fundamental que el clínico sospeche lo más precozmente posible el diagnóstico de FQ, por lo que debe ser incluido en el diagnóstico diferencial de cualquier cuadro respiratorio recurrente o persistente en los niños pequeños (Tabla 1).
Indicaciones del test del sudor
| • Síntomas respiratorios recurrentes (Neumonías o Sibilancias recurrentes, tos persistente) |
| • Diarrea crónica, mala absorción |
| • Retardo desarrollo pondoestatural |
| • Deshidratación hiponatrémica, hipoclorémica e hipokalémica con alcalosis metabólica |
| • Edema e hipoproteinemia |
| • Sabor salado de la piel |
| • IIeo meconial |
| • Hepatomegalia |
| • Prolapso rectal |
| • Pólipos nasales |
| • Ictericia prolongada del recién nacido |
| • Hermano con FQ |
Ref. 14: MINISTERIO DE SALUD, CHILE. Guía Clínica Fibrosis Quística Santiago: Minsal, 2007.
El standard de oro para establecer el diagnóstico sigue siendo el test del sudor. La muestra se obtiene de la piel del antebrazo mediante iontofo-resis con pilocarpina y se recolecta el sudor ya sea con gasa o papel filtro (técnica de Gibson y Cooke) o a través de microtúbulos (Macroduct). Se mide la concentración de cloro en la muestra y el diagnóstico se establece definitivamente con valores sobre 60 meq/L de cloro en 2 muestras consecutivas. Se considera sospechoso de la enfermedad cifras de 40 a 60 meq/L y normal bajo 40 meq/l.
Estudios recientes sugieren bajar el punto de corte de sospecha en el menor de 6 meses a 30 meq/L.
El test del sudor da cuenta del 98% de los casos de FQ, quedando un 2% de formas leves con examen límite o normal (6).
Los casos sospechosos deben repetir el test del sudor o realizar el estudio de mutaciones de FQ. La presencia de 2 mutaciones reconocidas como causantes de FQ situadas en posición trans constituye también diagnóstico definitivo.
La piel del recién nacido no permite una muestra adecuada de sudor, por lo que debe postergarse su realización hasta el mes de vida.
Falsos positivos aparecen en enfermedades poco frecuentes como displasia ectodérmica, hipotiroidismo, hipoparatiroidismo, fucosidosis, insuficiencia suprarrenal, SIDA, glucogenosis tipo I, mucopolisacárido-sis, deficiencia de glucosa 6 fosfatasa y desnutrición severa. Los falsos negativos son escasos y pueden verse en fallas de técnica y edema de la piel.
En los pocos casos en los que la duda diagnóstica persiste, se puede realizar la medición de la diferencia de potencial de la mucosa nasal, aprovechando la mayor electronegatividad que provoca la acumulación de cloro intracellular en los pacientes de FQ.
Pesquisa neonatalUno de los principales avances para mejorar el pronóstico de la enfermedad ha sido el desarrollo y la aplicación de la pesquisa neonatal para el diagnóstico precoz de la FQ. Esta consiste en tomar una muestra de sangre del talón del recién nacido y depositarla en un papel filtro, en el que se mide la concentración de la enzima Tripsina Inmunoreactiva, (IRT) que está elevada en el paciente FQ. Esto se hace en la misma tarjeta de Guthrie utilizada para la detección de TSH y PKU (pesquisa de hipotiroidismo y fenilquetonuria).
Hay distintos protocolos utilizados; todos parten tomando IRT en los primeros 3 días de vida. Aquellos por sobre el punto de corte (variable según el lugar de realización), repiten un segundo IRT a las 2 semanas (protocolo IRT/IRT) o analizan un set de DNA para las mutaciones más frecuentes del lugar, en la misma sangre del papel filtro inicial (protocolo IRT/DNA). De resultar positivo el segundo examen, se confirma el diagnóstico con el estándar de oro del test del sudor, a las 4 semanas de vida.
La sensibilidad de estos protocolos varía de 93 a 95% y el valor predicti-vo positivo es de 12,5% para IRT/IRT y de 15% para IRT/DNA.
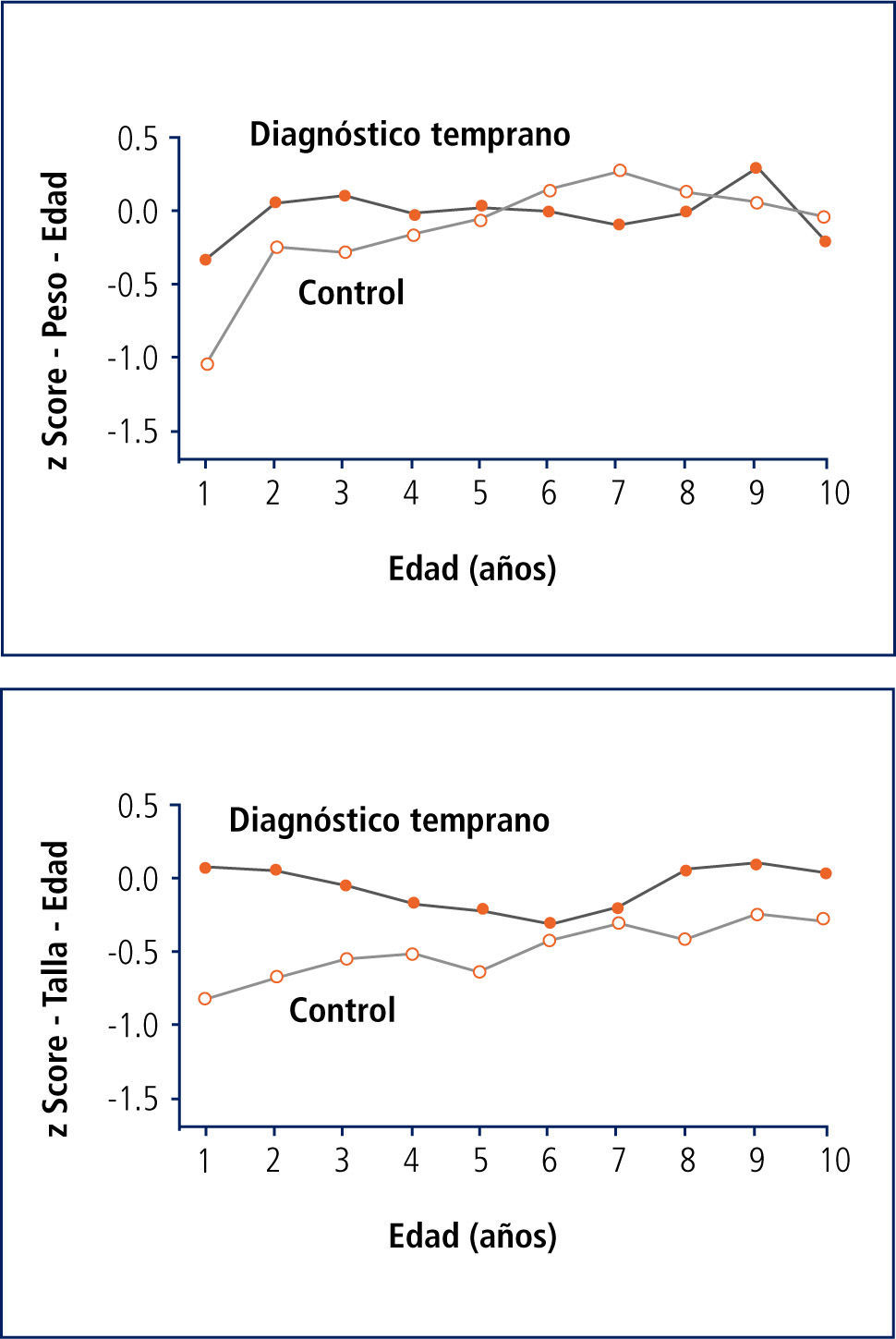
La experiencia de grandes cohortes con el diagnóstico neonatal data de 1985 en EE.UU., (Estudio Wisconsin) y dentro de las ventajas y beneficios se ha demostrado una mejoría significativa en la nutrición de los pacientes (Figura 6).

La mejor nutrición se asocia a menos infecciones pulmonares y por ende un mejor pronóstico.
El estudio de la cohorte randomizada de Wisconsin no demuestra una mejoría significativa de la función pulmonar en los lactantes diagnos- ticados precozmente por pesquisa, sin embargo datos de la cohorte holandesa muestran VEF1 estables v/s declinación entre pesquisa/controles y una cohorte histórica australiana revela un promedio de valores de VEF1 de 9,4% superior en el grupo diagnosticado por pesquisa.
Por último, revisiones sistemáticas del año 2006 muestran una mejoría significativa en sobrevida media a favor de los grupos diagnosticados por pesquisa neonatal (7 -11).
Avances en el tratamiento de la fqAvances en el tratamiento convencionalHasta el momento, el arsenal terapéutico para la FQ está dirigido a aliviar las consecuencias del defecto básico de la falla del CFTR, y conseguir así una mejor sobrevida y calidad de vida.
Los pilares de la terapia convencional en FQ siguen siendo la kinesite-rapia respiratoria, la mantención de la vía aérea libre de infección y la nutrición óptima. Estos pilares deben estar sustentados en un manejo multidisciplinario, sistemático y ordenado, donde el compromiso de los padres en el manejo diario y el compromiso de los médicos en la educación y el refuerzo positivo permanente a los niños y sus familia, son fundamentales.
1)Kinesiterapia respiratoriaEs básico el drenaje permanente de las secreciones bronquiales, desde el mismo día del diagnóstico, al menos dos veces al día, a lo largo de toda la vida del paciente con FQ. Los padres deben aprender las técnicas básicas y ser ellos los responsables de la KTR diaria.
Las técnicas varían con la edad del paciente:
- a)
Convencional en menores de 3 años:maniobras pasivas de presión, bloqueo, vibración y drenaje postural.
- b)
Convencional más técnica de espiración forzada:desde los 3 años, estimulando al niño a espirar suave y progresivamente para inducir el drenaje de las secreciones hacia la vía aérea central.
- c)
Convencional más drenaje autogénico:desde los 6 años, los niños aprenden maniobras autónomas de “ordeñe” de las secreciones desde las vías aéreas periféricas hacia las centrales.
Estas técnicas pueden complementarse con el uso del “Flutter”, que es una pipa que permite espirar contra una bola de acero induciendo movimientos oscilatorios hacia la vía aérea que desprenden las secreciones. La kinesiterapia puede apoyarse con el uso de mucolíticos, entre los cuales el mejor validado en la literatura es la Dnasa recombinante humana (Domase alfa, Pulmozyme), que hidroliza las cadenas del DNA, responsable de la alta viscocidad de las secreciones bronquiales. Se utiliza en nebulización a través de nebulizador jet con compresor de aire, en dosis de 2, 5mg(ampollas de 2mlde solución) una vez al día previo a la kinesiterapia.
2)AntibióticosEl control de la infección endobronquial es clave en la evolución del compromiso pulmonar de la FQ. La intervención precoz y agresiva de las complicaciones respiratorias son fundamentales en la mejoría de la sobrevida y el pronóstico de la FQ. El control mensual y permanente del cultivo de secreción bronquial permite detectar la primera infección por P. aeruginosa γ evitar el desarrollo de infección endobronquial crónica, objetivo principal del manejo médico. Se indican en diversas circunstancias:
a)Exacerbación agudaSe considera infección activa la presencia de cualquiera de las siguientes condiciones clínicas:
- •
Aparición o aumento de la tos;
- •
Aumento de la cantidad, viscosidad o coloración del esputo;
- •
Decaimiento, trastornos de la conducta habitual;
- •
Mal incremento ponderal, pérdida del apetito;
- •
Fiebre (pocas veces presente);
- •
Polipnea, dificultad respiratoria;
- •
Aumento de la signología pulmonar auscultatoria;
- •
Deterioro en las pruebas de función pulmonar.
En exacerbación debe iniciarse tratamiento antibiótico de acuerdo al germen presente en el cultivo de esputo y a la sensibilidad in vitro, eligiendo los agentes antibacterianos más adecuados. Debe considerarse siempre que la farmacodinámica de los antibióticos está aumentada en los pacientes de FQ, por lo que las dosis usadas son mayores que en los niños no FQ. Además, tratándose de una infección endobronquial, no debe esperarse cambios en la serie blanca ni elevación de parámetros de inflamación como VHS o proteína C reactiva, tanto para la indicación de antibióticos como para la duración del tratamiento.
Frente a infección por Pseudomonas aeruginosa, se debe asociar un be-talactámico con acción adecuada ante este germen, con un aminogli-cósido. Lo habitual es Ceftazidima 200mg/Kg/día en 4 dosis e.V., más Amikacina 20-30mg/Kg/día en 2 dosis e.V., con control de niveles plasmáticos del aminoglicósido para adecuar la dosis precisa. La duración del tratamiento no debe ser menor a 14 días. En exacerbaciones menores, sin signos de deterioro del estado general ni dificultad respiratoria, una buena alternativa es el uso de Ciprofloxacino oral en dosis de 40mg/Kg/ día en dosis por 14 días. Ciprofloxacino ha demostrado una excelente biodisponibilidad oral, adecuada concentración en el esputo, pero genera resistencia con rapidez y la cura bacteriológica es de menor duración.
Otras alternativas frente a resistencia bacteriana, son el Imipenem o el Meropenem, o la Ticarcilina, asociados siempre a aminoglicósido.
Frente a infección por Staphylococcus aureus, si es oxacilino sensible, de eleccición Cloxacilina sódica en dosis de 200mg/Kg/día en 4 dosis e.v. Alternativa en exacerbación menor, Flucloxacilina 100mg/Kg/día en 3 dosis oral. Si es oxacilino resistente, Vancomicina en dosis de 60mg/Kg/ día en 4 dosis e.v. mínimo de duración 21 días. Otras alternativas son Teicoplanina o la asociación Cotrimoxazol más Rifampicina oral. Frente a infección por Haemophylus influenzae, Cefotaxima 200mg/Kg/ día en 4 dosis e.V., o Amoxicilina/ácido clavulánico 100mg/Kg/día en 3 dosis oral, por 10 a 14 días.
b)En infección crónicaSe considera crónica la infección endobronquial, cuando los cultivos son persistentemente positivos por 6 meses o más, a pesar del uso adecuado de antibióticos. La experiencia danesa considera crónica una infección por P. aeruginosa cultivos positivos por menos de 6 meses pero con presencia de precipitinas para R. aeruginosa en el plasma.
En esta condición clínica se preconiza el uso de “curas periódicas” de antibióticos e.v. por 2 a 3 semanas cada 3 meses, sin esperar síntomas de exacerbación, lo que ha mejorado la sobrevida en estos casos.
c)Para evitar el desarrollo de infección crónicaLa sobrevida de los pacientes de FQ en Dinamarca tuvo un aumento sostenido desde que se implementó la política de tratar agresivamente la primera infección por R. aeruginosa, aún sin síntomas de exacerbación, intentando evitar el desarrollo de infección crónica. Para ello, indican una asociación de Colistín inhalatorio en dosis altas (1millón de unidades) asociado a Ciprofloxacino oral en dos dosis diarias por períodos de 3 semanas de uso alternados con 3 semanas de descanso.
d)Antibióticos inhalatoriosSe utilizan en FQ desde 1965, aunque con seguimientos prospectivos desde 1981.
Hay consenso de que frente a la primera infección endobronquial por R. aeruginosa la indicación terapéutica es antibióticos inhalados. Estos permiten obtener altas concentraciones del medicamento directo en la vía aérea, sin el riesgo de los efectos secundarios (oto y nefrotoxici-dad de los aminoglicósidos) y se han demostrado muy eficaces para erradicar la P. aeruginosa de la vía aérea. También se indican para el tratamiento de la infección endobronquial crónica, situación en la que son capaces de disminuir la velocidad de deterioro clínico y de caída de la función pulmonar, al disminuir la densidad bacteriana en el lumen de la vía aérea.
Los antibióticos inhalados aprobados por la evidencia médica hasta el momento son:
TOBI®: tobramicinaen solución para inhalación, en dosis de 300mg 2 veces al día, administrada a través de nebulizador PARI LC o eFLOW. Se administra durante 28 días, con 28 días de descanso, en meses alternos. La duración total de la terapia depende del objetivo clínico, desde 3 a 6 curas si se trata de erradicación de la primera infección por P. aeruginosa, hasta uso permanente con o sin antibióticos sistémicos cada 3 meses si se trata de mantener supresión bacteriana en los casos de infección endobronquial crónica.
Colistin inhalado: se usa en dosis de 1.000.000 unidades 2 veces al día asociado a Ciprofloxacino oral 20 - 30mg/kg/día en 2 dosis. Es antibiótico de elección en Dinamarca y otros países europeos. Se ha usado en curas de 3 semanas y hasta de 3 meses en casos de primera infección endobronquial por P. aeruginosa.
Recientemente se ha publicado la eficacia y seguridad de Aztreonam lisina para inhalación, en dosis de 75mg2 o 3 veces al día, demostrándose un claro efecto en mejoría de los síntomas, de la función pulmonar y para postergar la necesidad del uso de otros antibióticos anti Pseudomonas aeruginosa (12 -15,18).
MacrólidosSe ha probado que Azitromicina en dosis bajas (5mg/kg/día) y prolongadas tiene un efecto inmunomodulador y antiinflamatorio distinto al de antimicrobiano, que mejora la función pulmonar un promedio de 5,6% del VEF1, cifra similar al del efecto de la Dnasa. Se indica actualmente en pacientes con infección crónica por R. aeruginosa, y está en evaluación su uso en etapas mas precoces de la enfermedad (16, 17).
3)NutriciónFundamental para el pronóstico de la FQ es la mantención de una muy adecuada nutrición, ya que se ha demostrado una relación directa entre el estado nutricional y la función pulmonar. Esta relación implica que a mejor estado nutricional, mejor función pulmonar. Datos del grupo de FQ de Minessotta en EE.UU., en niños de 6 a 8 años, muestran que por cada kilo de ganancia de peso aumenta en 32 cc el VEF1.
La razón de este efecto es mutifactorial, probablemente el principal es que la malnutrición en edades tempranas altera el crecimiento y desarrollo del pulmón. Manteniendo una nutrición adecuada se posterga o evita el deterioro progresivo de la función pulmonar en los pacientes con FQ.
En un estudio comparativo de resultados de 2 centros de FQ, en Toronto y Boston, se encontró una sobrevida media de 30 años para el primero, y de 21 años para el segundo, con parámetros de función pulmonar comparables. La diferencia entre ambos centros está en los parámetros nutricionales, donde el grupo de Toronto ha establecido una dieta con ingesta no restringida en grasas.
El índice de masa corporal (IMC) ha demostrado ser el mejor parámetro de medición de la nutrición en pacientes de FQ, y el objetivo del manejo es mantener un IMC sobre el p25 para la edad, idealmente el p50.
Las recomendaciones nutricionales deben incluir:
- a)
Aporte calórico del 120 al 150% de la recomendación normal, sin restricción de grasas.
- b)
Enzimas pancreáticas en los casos en que se ha demostrado la malabsorción. Las más utilizadas son las cápsulas con microgránulos de cubierta entérica, (Ultrase®, Panzitrat®, Creón®) que vienen en dife rentes concentraciones de acuerdo a la cantidad de lipasa aportada. La dosis usual es de 2.000 U de lipasa por cada 120cc de fórmula o alimento materno para lactantes, 1.000 U/Kg por comida y 500 U/Kg para la colación en niños mayores. Las dosis deben ajustarse a la respuesta en ganancia de peso y los controles de la esteatorrea.
- c)
Aporte de vitaminas liposolubles, (A, D, E y K) al doble de la recomendación y elementos traza como Zn, Cu, Se, Ca y Fe (19).
Dos factores han incidido notablemente en la mejoría de la supervivencia media de los pacientes de FQ en los países desarrollados, que se relacionan con el “cómo” realizar el tratamiento convencional, y estos son:
Centros de fibrosis quísticaEs fundamental que los pacientes se incorporen a un manejo multidisciplina-rio integrado y sistemático, en un centro dedicado especialmente a la enfermedad. Las condiciones requeridas para formar un centro de FQ están delineadas en un consenso europeo y norteamericano del año 2005, e incluyen:
- •
Profesionales trabajando en equipo: neumólogo pediatra, gastro-enterólogo pediátrico, nutriólogo, kinesiólogo, endocrinólogo, genetista, infectólogo, psiquiatra y psicólogo, otorrino, cirujano digestivo y torácico, equipo de trasplante pulmonar, enfermera coordinadora el centro.
- •
Infraestructura:Test del sudor, departamento de imágenes, laboratorio de función pulmonar, laboratorio de microbiología, fibrobroncoscopía, salas de hospitalización y unidad de cuidados intensivos.
- •
Atención ambulatoria en consulta multidisciplinaria integrada, coordinada y registrada en base de datos.
- •
Un mínimo 50 pacientes en control.
- •
Debe tener un director responsable, habitualmente el neumólogo pediátrico (20).
Todas las complicaciones de la enfermedad deben estar escritas en protocolos de tratamiento sistematizados e incluir: protocolos de manejo: respiratorio, gastrointestinal, nutricional, endocrino y diabetes, cirugía pulmonar y digestiva, trasplante pulmonar.
En Chile, desde el año 2002 contamos con un Programa Nacional de FQ, desarrollado por el Ministerio de Salud, con financiamiento completo del programa, y con una guía clínica sistematizada del manejo de la FQ desde la sospecha, el diagnóstico y el manejo de todas las complicaciones de la enfermedad.
En la actualidad, la FQ está incorporada a las patologías con garantías explícitas de salud (GES), cuya cobertura asegura el acceso, la calidad, oportunidad y protección financiera de la atención de la enfermedad (12 -14).
Avances en nuevas terapias para la fqActualmente está en distintas fases de desarrollo la investigación dirigida a corregir el defecto básico, ya sea promoviendo la implantación del CFTR en la membrana plasmática o corrigiendo la alteración del líquido que debe cubrir la superficie epitelial (21).
Restauradores del líquido de la superficie epitelialSolución hipertónica de NaCI al 7%: está probado que la inhalación de solución de cloruro de sodio hipertónico logra aumentar la hidrata-ción de la vía aérea y mejora el clearance mucociliar, la función pulmonar y disminuye la frecuencia de las exacerbaciones (22).
Denufosol Tetrasodium: es un nuevo regulador de canal de iones, con acción multimodal, diseñado para corregir el defecto de transporte del CFTR, a través de estimular receptores P2Y2. Aumenta la secreción del ión cloro a través de un canal de cloro activado por calcio (Ca CC), e inhibe la absorción de sodio a través del canal de sodio (ENaC), además de aumentar la frecuencia del batido ciliar. Estas acciones integradas mejoran la hidratación del lumen y la depuración mucociliar.
Se ha probado seguridad y eficacia en estudios de fase II y se ha publicado recientemente un estudio clínico de fase III en mayores de 5 años con función pulmonar levemente alterada, en dosis de 60mg por vía inhalada 3 veces al día durante 24 semanas. Se demostró una mejoría significativa del VEF1 en relación al placebo (23, 24).
Bronchitol: es un manitol en polvo seco para administración inhalato-ria, ya se han completado estudios en fase II y III, demostrando también seguridad y eficacia, con mejoría significativa de la función pulmonar después de 2 semanas de uso (25).
Tratamientos dirigidos a corregir el defecto básico del cftrEstamos entrando en la era molecular de la terapia de la FQ, donde el tratamiento está basado en modular la falla del CFTR, por lo que son necesariamente mutación específicos. Ya hay numerosos estudios en fase II y III publicados, y algunos en fase III en pleno desarrollo, que ofrecen una expectativa de curación de la enfermedad en su defecto básico (26).
Moduladores para Mutaciones de Clase IEn este tipo de mutaciones no se produce CFTR por la presencia de codones de “stop prematuro” en el RNAm, también conocidas como mutaciones sin sentido, en las que los ribosomas no pueden leery transcribir la secuencia. El 10% de los casos de FQ corresponde a este tipo de mutaciones.
Ataluren (PTC 124), es una molécula que permite a los ribosomas leer la información genética “saltándose” los codones de “stop prematuro”. Acaba de publicarse un estudio de fase II, en la que se demuestra mejoría significativa de la diferencia de potencial nasal en niños, como reflejo de la aparición de CFTR en la superficie epitelial de la mucosa nasal. Está en desarrollo un estudio de fase III para evaluar resultados clínicos a corto y largo plazo (27).
Moduladores para Mutaciones de Clase IIVX-809: es una molécula “correctora” de CFTR, que permite su tránsito a través del aparato de Golgi evitando la degradación de la proteína en su camino hacia la superficie celular. Estudios in vitro demuestran un aumento de 3 a 15% de la cantidad de CFTR en membranas celulares con p508del (28).
Moduladores para Mutaciones de Clase IIIVX-770: es una molécula “potenciadora” de CFTR, que permite superar el “bloqueo” del canal. Se ha probado su uso clínico en la mutación específica G551D, observándose una disminución significativa de la concentración de cloro en el sudor y también de la diferencia de potencial en la mucosa nasal (29).
Está en desarrollo actualmente un estudio fase III en niños y adultos, que evalúa el uso combinado de VX-770 y VX-809.
Aún falta largo camino por recorrer con este tipo de terapias moleculares, falta aún por evaluar sus efectos benéficos y efectos secundarios a largo plazo. De todos modos, tienen la potencialidad de alterar el curso de la enfermedad pulmonar, por lo que debieran aplicarse lo más precozmente posible, antes de que se inicie el deterioro de la función pulmonar y constituyen una esperanza de mejoría del pronóstico y de la calidad de vida de nuestros pacientes de FQ.
ConclusionesLa FQ continua siendo un gran desafío de diagnóstico y terapéutico en nuestro medio.
Son tareas pendientes para nuestro país el diagnóstico y la intervención precoz de la FQ, a través del establecimiento de la pesquisa neonatal y el desarrollo de un Centro de FQ donde el paciente y su familia encuentren un manejo multidisciplinario, sistemático y protocolizado.
Las investigación de nuevas terapias dirigidas a corregir el trastorno fi-siopatológico básico o directamente a modular las distintas mutaciones del CFTR, abren una luz de esperanza de conseguir una supervivencia cada vez mayor y, sobre todo, de asegurar una calidad de vida comparable a lo normal, en nuestros pacientes de FQ
El autor declara no tener conflictos de interés, en relación a este artículo.