




La evaluación de la susceptibilidad hereditaria al cáncer se considera un estándar de atención, ya que afecta no sólo al clínico en la comprensión de la causa del cáncer, sino también, a las opciones de prevención y tratamiento. Las raíces de nuestro conocimiento sobre los síndromes hereditarios del cáncer se pueden remontar a informes tempranos de familias con historias notables de cáncer. El objetivo de este artículo es revisar la cronología histórica de los dos síndromes de cáncer hereditario más comúnmente evaluados, cáncer de mama y ovario hereditario y el síndrome de Lynch. Aunque muchas personas identificadas con estos síndromes hoy provienen de familias similares a las reportadas en los primeros informes históricos, nuestra comprensión de estos síndromes, su expresión y penetración, ha evolucionado tras los años. Además, el aumento de la utilización de amplios paneles multi-gen sigue añadiendo información a la complejidad de la definición de fenotipos asociados. Estos hallazgos pueden ser un desafío en la traducción de los resultados al manejo clínico para los pacientes y las familias, pero también proporcionan una oportunidad para ganar comprensión de los fundamentos genéticos de la etiología del cáncer.
La evaluación de la susceptibilidad hereditaria al cáncer se ha convertido en parte integral de la oncología, impactando no sólo en la comprensión por parte de los clínicos de la causalidad del cáncer, si no también influenciando en las opciones de atención preventiva y tratamiento. La amplia integración de la genética del cáncer en la atención oncológica ha seguido creciendo, impulsado por los avances en la secuenciación de genes, que ha reducido los costos. Las raíces de nuestro conocimiento actual sobre los síndromes de cáncer hereditario se remontan a los primeros informes médicos y científicos de familias con excepcionales historias de cánceres de inicio precoz, afectando a múltiples generaciones o afectando a individuos con fenotipos específicos. El propósito de este artículo es revisar la historia de los dos síndromes de cáncer hereditario más conocidos: el síndrome cáncer hereditario de mama y ovario (HBOC por “hereditary breast and ovarian cancer sydnrome”) y el síndrome de Lynch (LS por “Lynch syndrome”). Estos síndromes no fueron los primeros por son lo más comunes. Aunque las recomendaciones y pautas de detección y manejo pueden variarse entre las organizaciones profesionales, se resume las directrices corrientes de la Red Nacional de Centros Integrales de Cáncer en Estados Unidos (EE.UU.) de Norteamérica (NCCN por “National Comprehensive Cancer Center Network”) para estos síndromes en la Tabla 1 y Tabla 2.
RESUMEN DE LAS GUÍAS DE MANEJO DE LOS SÍNDROMES DE MAMA DE NCCN
| CÁNCER DE MAMA | CÁNCER DE OVARIO | OTROS |
|---|---|---|
| MUJER A partir de los 25 años • Examen clínico, cada 6-12 meses A partir de los 25-29 años • Resonancia magnética con contraste (RMN) anualmente o mamografía si la RMN no está disponible De los 30-75 años • Mamografía y RMN con contraste anual Mayor de los 75 años • Gestión considerada individualmente Discutir la opción de la mastectomía preventiva Considere los agentes reductores del riesgo HOMBRE A partir de los 35 años • Entrenamiento de auto-examen • Examen clínico, anualmente | Salpingo-ooforectomía bilateral preventiva • A los 35-40 años, después de tener los hijos deseados • Podría esperar en los portadores de mutaciones en BRCA2 hasta los 40-45 años, debido a un edad de diagnóstico más tardía No se ha demostrado que la ecografía transvaginal y la CA-125 sean lo suficientemente sensibles o específicas para una recomendación de cribado, pero pueden considerarse, de 30 a 35 años de edad, a la discreción del médico Considere los agentes reductores del riesgo | Cáncer de la próstata: A partir de los 45 años • Recomendar la detección en los portadores mutaciones en BRCA2 • Considerar la detección en los portadores de mutaciones en BRCA1 Cáncer de páncreas y la melanoma: • Individualizado basado en la historia familiar |
National Comprehensive Cancer Center Network (NCCN) Genetic/Familial High-Risk Assessment: Breast and Ovarian, version 2.2017
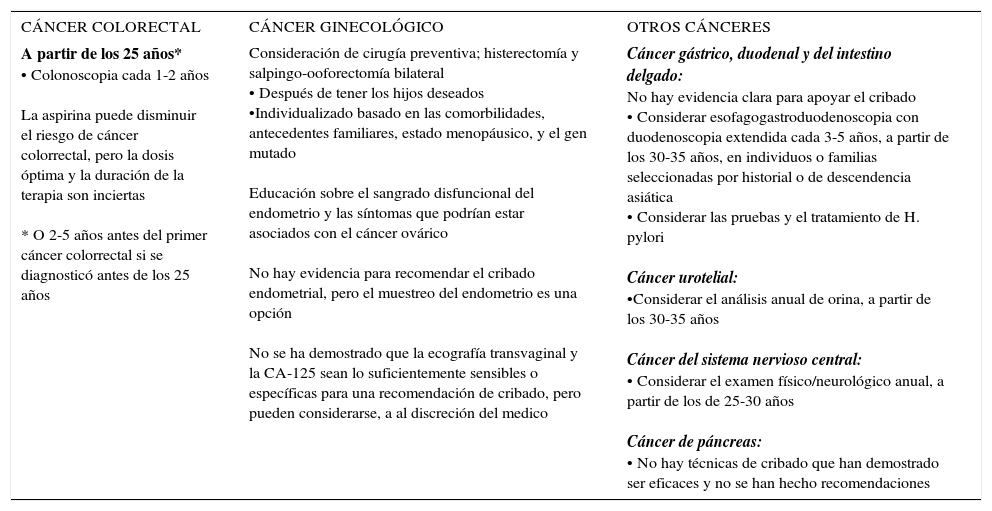
RESUMEN DE LAS GUÍAS DE MANEJO DEL LS DE NCCN
| CÁNCER COLORECTAL | CÁNCER GINECOLÓGICO | OTROS CÁNCERES |
|---|---|---|
| A partir de los 25 años* • Colonoscopia cada 1-2 años La aspirina puede disminuir el riesgo de cáncer colorrectal, pero la dosis óptima y la duración de la terapia son inciertas * O 2-5 años antes del primer cáncer colorrectal si se diagnosticó antes de los 25 años | Consideración de cirugía preventiva; histerectomía y salpingo-ooforectomía bilateral • Después de tener los hijos deseados •Individualizado basado en las comorbilidades, antecedentes familiares, estado menopáusico, y el gen mutado Educación sobre el sangrado disfuncional del endometrio y las síntomas que podrían estar asociados con el cáncer ovárico No hay evidencia para recomendar el cribado endometrial, pero el muestreo del endometrio es una opción No se ha demostrado que la ecografía transvaginal y la CA-125 sean lo suficientemente sensibles o específicas para una recomendación de cribado, pero pueden considerarse, a al discreción del medico | Cáncer gástrico, duodenal y del intestino delgado: No hay evidencia clara para apoyar el cribado • Considerar esofagogastroduodenoscopia con duodenoscopia extendida cada 3-5 años, a partir de los 30-35 años, en individuos o familias seleccionadas por historial o de descendencia asiática • Considerar las pruebas y el tratamiento de H. pylori Cáncer urotelial: •Considerar el análisis anual de orina, a partir de los 30-35 años Cáncer del sistema nervioso central: • Considerar el examen físico/neurológico anual, a partir de los de 25-30 años Cáncer de páncreas: • No hay técnicas de cribado que han demostrado ser eficaces y no se han hecho recomendaciones |
National Comprehensive Cancer Center Network (NCCN) Genetic/Familial High-Risk Assessment: Colorectal, version 1.2017
En 1866, un cirujano francés, Pierre Paul Broca describió la historia de cáncer de mama en la familia de su esposa, que incluyó a diez mujeres con cáncer de mama en más de tres generaciones 1,2. Desde entonces, muchos investigadores han continuado documentando antecedentes de mujeres con cáncer de mama y su historia familiar, así como de familias con historias notables de cáncer de mama 3–7. En 1988, Newman y cols. describieron los resultados de sus análisis de segregación, proponiendo un modelo autosómico dominante para el cáncer de mama con un alelo de susceptibilidad altamente penetrante 8. Pocos años después, el equipo de la Dr. Mary Clare King en la Universidad de California, Berkeley, identificó un locus en el cromosoma 17 usando análisis de ligamiento en familias con cáncer de mama precoz 9.
El Dr. Steven Narod, parte del equipo de investigadores de la Universidad Mc Gill en Montreal, publicó sobre varias familias con cáncer de mama y de ovario con ligamiento positivo al mismo locus 10. Aceleró una búsqueda internacional para localizar el gen causante en este locus y en 1994 fue localizado el gen supresor de tumores BRCA111. En el mismo año, investigadores del Institute of Cancer Research en el Reino Unido publicaron sobre sus esfuerzos colaborativos para identificar un segundo locus de cáncer de mama, BRCA2, en el cromosoma 13 12,13.
El síndrome de cáncer hereditario de mama y ovario (HBOC) causado por mutaciones en los genes BRCA, se caracteriza por cáncer de mama precoz y presencia del cáncer ovárico, pero individuos que portan mutaciones también corren riesgo para otros tipos de cáncer, como el cáncer de mama masculino, cáncer de próstata, cáncer de páncreas y melanoma 14–17. Las primeras estimaciones de riesgo de cáncer de mama, en portadoras de mutaciones en BRCA1 o BRCA2 señalaban riesgo superior al 80% a lo largo de la vida 18–20. Sin embargo, estudios posteriores han evidenciado menores riesgos acumulativos de cáncer de mama, del orden de 40-50% 17,21–23. De modo similar, los primeros estudios estimaron el riesgo cáncer de ovario, de 63% para portadoras de BRCA1 y de 27% para BRCA218,19 pero estudios más recientes han proporcionado estimaciones de menor riesgo, del orden de 39-59% para el BRCA1 y de 17-21% para el BRCA217,21–23.
Años de investigación proporciona una mejor comprensión de las características patológicas e histológicas de los cánceres asociados con mutaciones en los genes BRCA. Los cánceres de mama asociados con BRCA1 tienen más probabilidades de tener una histología de alto grado y medular y de ser triples negativos (caracterizado por la ausencia de la expresión de los receptores de estrógeno, de progesterona y HER2). Los cánceres ováricos identificados en los portadores de mutación BRCA tienden a ser adenocarcinomas serosos de alto grado.
Mutaciones en los genes BRCA1 y BRCA2 fueron identificadas inicialmente en familias de cáncer de mama y ovario muy penetrantes, seleccionadas debido a su marcado historial personal y familiar. Gran parte de la investigación original sobre penetrancia, expresión y riesgo se derivó de estas familias seleccionadas. Sin embargo, las directrices que dirigen cuando iniciar un análisis de los genes BRCA1 y BRCA2, son más amplias en hoy día y han llevado a una comprensión expandida del fenotipo de HBOC.
Por ejemplo, el NCCN recomienda efectuar análisis de BRCA1 y BRCA2 en todas las mujeres con cáncer de mama que debute antes de los 45 años, en todas las mujeres con cáncer de mama triple negativo bajo los 60 años y en todas las pacientes con cáncer de ovario epitelial 24. Además, define criterios adicionales para otros cánceres relacionados con BRCA, así como recomienda realizar análisis a familias que no han presentado estos cánceres a edad temprana, pero tienen múltiples miembros de la familia, diagnosticados con cáncer 24.
La historia científica de BRCA1 y BRCA2 está siempre entretejida con la historia legal del patentamiento de genes. Aunque la publicación original, la identificación de los loci BRCA1 y BRCA2 fueron publicados por otros grupos, investigadores de la Universidad de Utah, publicó las secuencias completas de ambos genes 11,25. Simultáneamente, estos investigadores, liderados por el Dr. Mark Skolnick, solicitaron la protección de patentes de las formas aisladas de BRCA1 y BRCA2, los primers y sondas usadas para secuenciar ambos genes, y los métodos utilizados 26. Las patentes se asignaron a ellos y después licenciadas a Myriad Genetics, una compañía de biotecnología fundada en 1991 por los Dres. Mark Skolnick y Peter Meldrum. Después de que las patentes fueron emitidas, Myriad Genetics aplicó activamente sus patentes contra los investigadores clínicos, y se convirtió en el único laboratorio comercial para ofrecer pruebas BRCA1 y BRCA2. Estas patentes permanecieron sin cuestionar hasta 2009, cuando la Unión Americana de Libertades Civiles (ACLU por el “American Civil Liberties Union”) presentó una demanda contra Myriad Genetics en nombre de pacientes, clínicos, investigadores, grupos de defensa y organizaciones científicas, en una corte federal en Nueva York en los EE.UU. El caso pasó de la corte federal, la Corte de Apelaciones de los EE.UU, y finalmente la Corte Suprema. En junio de 2013, la Corte Suprema sostuvo que “un segmento natural de ADN es un producto de la naturaleza y no es elegible para patentes simplemente porque ha sido aislado, pero que el ADNc es elegible porque no es natural”. Con este fallo, el monopolio de Myriad sobre las pruebas BRCA1 y BRCA2 terminó y otros competidores entraron en el mercado, impactando significativamente el panorama de las pruebas genéticas.
El fallo de la Corte Suprema sobre las patentes genéticas de BRCA llegó dentro de un mes del editorial New York Times de Angelina Jolie titulado “My Medical Choice” (Mi Elección Médica). En esta noticia, narra su experiencia con la prueba genética de BRCA1 y su decisión de someterse a una mastectomía bilateral profiláctica después de salir “positiva” por la mutación. La celebridad internacional de la Sra. Jolie impulsó su editorial y el impacto en la conciencia pública ha sido estudiado. Este impacto se ha denominado “Angelina Jolie Effect” (Efecto Angelina Jolie) para referirse al aumento de la conciencia de las pruebas genéticas, la búsqueda de información sobre pruebas y prevención y el interés en las pruebas genéticas que siguieron 27–29.
SÍNDROME DE LYNCH (LS)En 1913, el Dr. Aldred Scott Warthin, PhD, Director del Departamento de Patología de la Universidad de Michigan en Ann Arbor, EE.UU., reportó los resultados de una revisión de los registros médicos de pacientes tratados en la Universidad de Michigan, acerca de cuatro familias con historias sorprendentes de cáncer multi-generacional 30. Una de estas familias, pertenecientes a la costurera del Dr. Warthin (mujer que posteriormente falleció de cáncer de endometrio), es conocida como “familia de G”, que fue estudiada exhaustivamente por décadas y se destacó por la predominancia de cáncer uterino, cáncer gástrico y cáncer intestinal 30–34. En 1966, el Dr. Henry Lynch reportó acerca de dos grandes familias de Nebraska y Michigan con un espectro similar de cánceres 35.
Lynch y cols. propusieron que una herencia autosómica dominante era la etiología más probable para el cáncer en estas familias, caracterizada por múltiples generaciones de cáncer colorrectal de aparición temprana sin poliposis 35. El término cáncer colorrectal hereditario sin poliposis (HNPCC por Hereditary non-polyposis colorectal cancer) fue propuesto para distinguir las familias de cáncer colorrectal con pólipos, de las personas con poliposis adenomatosa familiar 36–38. Sin embargo, los términos síndrome de Lynch I y síndrome de Lynch II también fueron utilizados para distinguir un fenotipo de familias con historias específicas de cáncer colorrectal (LS I), de las personas con cáncer colorrectal y otro tipo de cáncer, especialmente endometrial (LS II) 39–41.
Para identificar a las familias con un diagnóstico clínico de LS, el grupo colaborativo internacional sobre cáncer colorrectal hereditario sin poliposis, estableció algunos criterios en 1991. Estos son conocidos como criterios Amsterdam I, que requieren por lo menos tres parientes con cáncer colorrectal; uno de los cuales debe ser pariente de primer grado y con al menos dos generaciones sucesivas afectadas con cáncer, así como al menos un miembro de la familia diagnosticado antes de los 50 años 42. Sin embargo, los criterios Amsterdam I no representarían adecuadamente otros cánceres asociados a LS, limitando la capacidad de identificar a las familias con LS. Los criterios de Amsterdam II se formularon para permitir otros cánceres asociados extra colónicos en las familias LS 43.
En 1993, se describió el primer gen MSH244,45, un gen de reparación de errores de replicación (MMR por “mismatch repair”), ligado a LS, seguido el año siguiente por la descripción del gen MLH146,47 y del gen PMS248 y en 1997, por el descubrimiento del gen MSH653. En el año 2009, se reportaron deleciones en los últimos exones del gen EpCam (epitelial cell adhesión molecule) situado al lado del MSH2 y que fueron implicados en el desarrollo de LS 49. Los estudios moleculares iniciales de LS demostraron que la mayoría de las familias tenían mutaciones en MLH1 y MSH2. Sin embargo, las pruebas genéticas más amplias han demostrado que las mutaciones en MSH6 y PMS2 han sido subestimadas.
Aunque las estimaciones de riesgo de cáncer varían según el gen, los individuos con LS tienen riesgo elevado de desarrollar cáncer colorrectal, cáncer de endometrio, cáncer gástrico, cáncer de ovario, así como de intestino, hépato-biliares, vías urinarias, cerebro y piel. Los primeros estudios de familias que cumplían los criterios clínicos para LS, demostraron sobre un 80% de riesgo de cáncer colorrectal a lo largo de la vida 50. Estudios más recientes de portadores de la mutación en los genes MLH1 y MSH2 han estimado que las mujeres tienen un 43-52% de riesgo de cáncer colorrectal y los hombres un riesgo de 66-69% a lo largo de su vida 51,52. El riesgo de cáncer colorrectal asociado con una mutación en MSH6ha mostrado ser 20% para las mujeres y 44% para los hombres 53. El riesgo de cáncer colorrectal para los portadores de una mutación en el gen PMS2 se estima que 15-20% 54. Las mujeres con LS también tienen un riesgo de 25-60% de desarrollar cáncer de endometrio 50,55. El riesgo de cáncer gástrico es 6-13% y el riesgo de cáncer de ovario puede llegar a cifras de 11% 56.
Más allá del fenotipo de cáncer definido, se identificaron características patológicas distintas de tumores asociados con LS. Los cánceres de colon en individuos con LS son más frecuentemente de lado derecho y tienen rasgos histológicos característicos, con tendencia a ser mucinoso y mal diferenciados, más linfocitos infiltrantes de tumores. Otra característica de estos tumores es la inestabilidad microsatélital (MSI). Los microsatélites son repeticiones en tándem en el ADN, generalmente de uno a cinco pares de bases de largo, que se encuentran a lo largo del genoma. La inestabilidad microsatélite se refiere a un número anormal de estas repeticiones en un tumor, ya sea una reducción o expansión, en comparación con el tejido normal. Esta asociación de inestabilidad MSI con el cáncer de colon familiar se reportó en 1993, en dos publicaciones en la misma edición de Nature, señalando que inestabilidad MSI se correlacionó con los tumores en el colon proximal 57 y se sobrerrepresentó en los tumores de los casos familiares, en comparación con el cáncer colorrectal esporádico 58. Hallazgos similares se replicaron más tarde ese año en individuos con cánceres endometriales esporádicos y familiares, lo que demostró que la inestabilidad MSI era más común entre los casos familiares 59. Aunque no todos los cánceres colorrectales y endometriales con inestabilidad MSI son atribuibles a LS, la mayoría de éstos en individuos con LS serán inestables, haciendo que la inestabilidad MSI sea un indicador importante de la etiología del cáncer. El cáncer colorrectal esporádico que muestra instabilidad de MSI puede atribuirse a la hipermetilación adquirida del gen MLH1.
En 1997, las guías de Bethesda fueron desarrolladas y actualizadas en 2004, para identificar a cuáles pacientes de cáncer colorrectal deberían hacer estudios MSI en sus tumores 60,61.
Estas guías definieron cinco criterios:
- 1)
Cáncer colorrectal diagnosticado menor de los 50 años,
- 2)
Múltiples cánceres colorrectales primarios u otros tumores asociados con LS, independientemente de la edad del diagnóstico,
- 3)
Cáncer colorrectal con histología MSI-alta diagnosticada menor de los 60 años,
- 4)
Cáncer colorrectal diagnosticado en uno o más parientes de primer-o segundo-grado con un cáncer asociado con LS, uno diagnosticado menor de los 50 años ó
- 5)
Cáncer colorrectal diagnosticado en dos o más parientes de primer-o segundo-grado con tumores asociados a LS, independientemente de la edad del diagnóstico.
En 2008 Hampel y col. publicaron de una serie de pacientes con cáncer colorrectal a cuyos tumores hicieron estudios de MSI y inmunohistoquímica de las proteínas MMR (IHQ). En aquellos con estudios tumorales anormales hicieron análisis genéticos de los genes MMR y encontraron que el 3.6% tenían LS. Más notable, es que el 28% de estos individuos no se habrían identificado si los estudios de tumores se hubieran limitado a los que cumplían con las guías de Bethesda 62. En 2009, el Grupo de Trabajo de Evaluación de las Aplicaciones Genómicas en la Práctica y la Prevención (EGAPP por “Evaluation of Genomic Applications in Practice and Prevention”) encontró que había evidencia suficiente para recomendar la evaluación para LS en todos los pacientes con cáncer colorrectal recientemente diagnosticados 63. El cribado universal de los tumores, de pacientes con cáncer colorrectal y endometrial, ya sea por MSI o IHC de los proteínas MMR, ha tenido éxito variable en su implementación, pero está claro que la utilización de criterios de Amsterdam o guías Bethesda, por sí solos, fallará en identificar una proporción significativa de familias con LS. Por lo tanto, se recomiendan criterios más amplios, en conjunto con pruebas de tumores, para identificar familias con LS 63–65.
Varios estudios publicados en 2014, añadieron evidencia para la comprensión de la etiología de la deficiencia en la reparación del mal emparejamiento en cánceres colorrectal y endometrial 66–68. Estos estudios demostraron que el 52-69% de los tumores que tienen deficiencia de reparación inexplicable, son atribuibles a múltiples mutaciones somáticas de los genes MMR o por la pérdida de heterozigosidad (LOH) en el tumor. Esto representa un avance significativo, ya que en el pasado, individuos con tumores que demostraban deficiencia de la reparación inexplicables, especialmente la pérdida de MSH2 y MSH6 por IHQ, que se manejaban como LS, a pesar de no tener una mutación germinal detectable.
CONCLUSIÓNHistóricamente, los síndromes de tumores hereditarios revisados en este artículo, fueron identificados utilizando pruebas genéticas dirigidas y orientadas por los diagnósticos diferenciales basados en características del fenotipo de los pacientes. Por lo tanto, las pruebas genéticas son altamente selectivas y orientadas por nuestro conocimiento del fenotipo. Esto queda ejemplificado en las diferencias en las primeras estimaciones de desarrollar cáncer en las familias con mutaciones del gen BRCA, que resultaron de estudios en familias altamente seleccionadas por su cercanía, en comparación con las ulteriores estimaciones que señalaban menor riesgo, que fueron generadas a partir de cohortes con parentescos mucho más amplios. Hoy, la mayor utilización de pruebas con amplios paneles multi-genes, permite la evaluación simultánea de varios genes y muchos paneles también incluyen genes que no pueden ser parte de los que se incluyen en el diferencial. Como tal, existe una creciente información sobre la identificación de mutaciones de alto riesgo que portan individuos con diagnóstico de cáncer, no tradicionalmente asociado con mutaciones de línea germinal. Aunque estos resultados plantean desafíos con la transferencia de este conocimiento a la práctica clínica, nuestra comprensión de cómo hemos definido estos síndromes en el pasado, servirá de experiencia para facilitar estas nuevas definiciones en el futuro.
La autora declara no tener conflictos de interés en relación a este artículo.