




Assessment for hereditary susceptibility to cancer is considered standard of care, as it impacts not only a clinician's understanding of cancer causation but also options for prevention and treatment. The roots of our current knowledge about hereditary cancer syndromes can be traced to early reports of families with striking cancer histories. The purpose of this article is to review the historical timeline of the two most commonly assessed hereditary cancer syndromes, hereditary breast and ovarian cancer (HBOC) and Lynch syndrome (LS). While many individuals identified with these syndromes today come from families similar to those seen in the early historical reports, our understanding of these syndromes, their expression and penetrance, has evolved over the years. In addition, the increased utilization of broad multi-gene panels continues to add to the complexity of defining associated phenotypes. These findings can lead to challenges with translating results to clinical management for patients and families, but also provide an opportunity to continue to gain understanding of the genetic underpinnings of cancer etiology.
Today, assessment for hereditary susceptibility to cancer has become an integral part of oncology, impacting not only a clinician's understanding of cancer causation but also influencing treatment and preventive care options. The widespread integration of cancer genetics into oncologic care has continued to grow, fueled by sequencing advances that have reduced cost. The roots of our current knowledge about hereditary cancer syndromes can be traced to early medical and scientific reports of families with striking histories of early-onset cancers over multiple generations or individuals with specific phenotypes. The purpose of this article is to review the historical timeline of the two most commonly assessed hereditary cancer syndromes, hereditary breast and ovarian cancer (HBOC) and Lynch syndrome (LS). These syndromes were not the first hereditary cancer syndromes to be defined, nor were the genes associated the first to be discovered. However, based on our current understanding, they are the most common and the basis of much of genetic testing that is performed today. While screening and management recommendation/guidelines vary by professional organization and, a summary of current NCCN (National Comprehensive Cancer Center Network) guidelines for these syndromes is provided in Table 1 and Table 2.
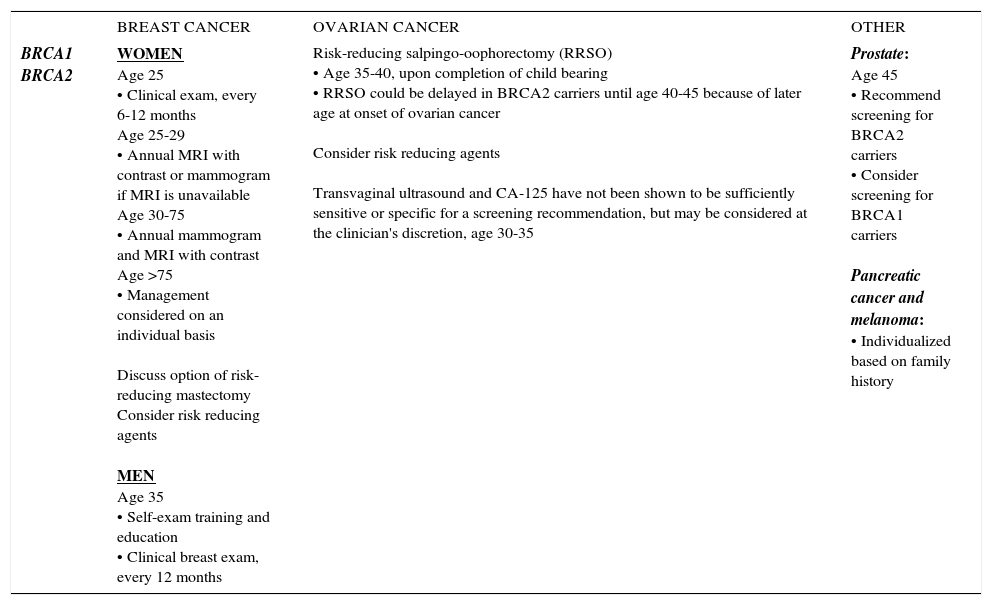
SUMMARY OF NCCN HBOC MANAGEMENT GUIDELINES, 2017
| BREAST CANCER | OVARIAN CANCER | OTHER | |
|---|---|---|---|
| BRCA1 BRCA2 | WOMEN Age 25 • Clinical exam, every 6-12 months Age 25-29 • Annual MRI with contrast or mammogram if MRI is unavailable Age 30-75 • Annual mammogram and MRI with contrast Age >75 • Management considered on an individual basis Discuss option of risk-reducing mastectomy Consider risk reducing agents MEN Age 35 • Self-exam training and education • Clinical breast exam, every 12 months | Risk-reducing salpingo-oophorectomy (RRSO) • Age 35-40, upon completion of child bearing • RRSO could be delayed in BRCA2 carriers until age 40-45 because of later age at onset of ovarian cancer Consider risk reducing agents Transvaginal ultrasound and CA-125 have not been shown to be sufficiently sensitive or specific for a screening recommendation, but may be considered at the clinician's discretion, age 30-35 | Prostate: Age 45 • Recommend screening for BRCA2 carriers • Consider screening for BRCA1 carriers Pancreatic cancer and melanoma: • Individualized based on family history |
National Comprehensive Cancer Center Network (NCCN) Genetic/Familial High-Risk Assessment: Breast and Ovarian, version 2.2017
SUMMARY OF NCCN LYNCH SYNDROME MANAGEMENT GUIDELINES, 2017
| COLORECTAL CANCER | GYNECOLOGIC CANCERS | OTHER |
|---|---|---|
| Age 20-25* • Colonoscopy every 1-2 years Aspirin may decrease the risk of colorectal cancer, but optimal dose and duration of therapy are uncertain *Or 2-5 years earlier than the youngest cancer diagnosed, if before age 25 | Consider risk-reducing salpingo-oophorectomy (RRSO) upon completion of child bearing Education about dysfunctional endometrial bleeding and the symptoms that are associated with ovarian cancer There is no clear evidence to support endometrial screening, but annual endometrial sampling is an option Transvaginal ultrasound and CA-125 have not been shown to be sufficiently sensitive or specific for ovarian cancer screening, but may be considered at the clinician's discretion | Gastric, duodenal, and small bowel cancer: • No clear evidence to support screening • Consider esophagogastroduodenoscopy with extended duodenoscopy every 3-5 years, age 30-35, in selected individuals or families or those of Asian descent • Consider testing and treating H. pylori Urothelial cancer: Consider annual urinalysis, age 30-35 Central nervous system cancers: Consider annual physical/neurologic exam, age 25-30 Pancreatic cancer: • No effective screening techniques available or recommendations have been made |
National Comprehensive Cancer Center Network (NCCN) Genetic/Familial High-Risk Assessment: Colorectal, version 1.2017
In 1866, French surgeon, Pierre Paul Broca described the history of breast cancer in his wife's family which included ten women with breast cancer over three generations 1,2. Researchers continued to document studies of women with breast cancer and family history, as well as families with striking breast cancer histories 3–7. In 1988, Newman et al described the results of their segregation analysis which proposed an autosomal dominant model for a highly penetrant susceptibility allele for breast cancer 8. Within a few years, Dr. Mary Clare King's group at the University of California, Berkeley, identified a locus on chromosome 17 using linkage analysis in families with early onset breast cancer 9. Dr. Steven Narod, part of a group of researchers at Mc Gill University in Montreal, reporting on several families with both breast and ovarian cancer with positive linkage to the same locus 10. There was a subsequent international quest to identify the causative gene at this locus and in 1994, the BRCA1 gene was localized 11. In the same year, researchers from the Institute of Cancer Research in the United Kingdom published on their collaborative efforts identifying a second breast cancer locus, BRCA2, to chromosome 13 12,13.
Hereditary breast and ovarian cancer (HBOC), caused by mutations in the BRCA1 and BRCA2 genes is typified by early-onset breast cancer and ovarian cancer, but individuals are also at elevated risk for other cancers, including male breast cancer, prostate cancer, pancreatic cancer, and melanoma 14–17. Early risk estimates of breast cancer in BRCA mutation carriers estimate lifetime risk to be over 80% 18–20. However, later studies have supported lower cumulative breast cancer risks of 40-50% 17,21–23. Similarly, early studies estimated ovarian cancer risk to be 63% and 27%, respectively, for BRCA1 and BRCA218,19 with more recent studies providing lower estimates of 17-21% for BRCA2 and 39-59% for BRCA117,21–23. BRCA1 associated breast cancers are more likely to be triple-negative (negative for estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2), high-grade and medullary histology. The ovarian cancers identified in BRCA mutation carriers tend to be high-grade serous adenocarcinomas.
BRCA1 and BRCA2 mutations were initially identified in highly penetrant breast and ovarian cancer families, selected because of their striking personal and family histories. Much of the original research about penetrance, expression, and risk was derived from these selected families. However, current, broader testing guidelines, have led to a more expanded understanding of the HBOC phenotype. For example, the National Comprehensive Cancer Center Network (NCCN) recommends analysis of BRCA1 and BRCA2 in all women with breast cancer age 45 and under, all women with triple-negative breast cancer age 60 and under, and all epithelial ovarian cancer patients 24. Additional criteria account for other BRCA related cancers, as well as families with multiple closely related family members diagnosed, regardless of age at diagnosis 24.
The scientific story of BRCA1 and BRCA2 is forever interwoven with the legal history of gene patenting. While the original publication, identifying the BRCA1 and BRCA2 loci were published by other groups, researchers from the University of Utah, published the complete sequences of both genes 11,25. Concurrently, these researchers, lead by Dr. Mark Skolnick applied for patent protection of the isolated forms of BRCA1 and BRCA2, the primers and probed used to sequence both genes, and the methods utilized 26. The patents were assigned and then licensed to Myriad Genetics, a biotechnology company founded in 1991 by Drs. Mark Skolnick and Peter Meldrum. After the patents were issued, Myriad Genetics actively enforced their patents against clinical researchers, and became the sole commercial laboratory to offer BRCA1 and BRCA2 testing. These patents remained unchallenged until 2009, when the American Civil Liberties Union (ACLU) filed suit against Myriad Genetics on behalf patients, clinicians, researchers, advocacy groups and scientific organizations, in federal court in New York. The case moved from the federal court, the United States Court of Appeals, and finally the Supreme Court. In June of 2013, the Supreme Court held that “a naturally occurring DNA segment is a product of nature and not patent eligible merely because it has been isolated, but that cDNA is patent eligible because it is not naturally occurring.” With this ruling, Myriad's monopoly on BRCA1 and BRCA2 testing ended and other laboratory competitors entered the market, significantly impacting the landscape of BRCA1 and BRCA2 testing.
The Supreme Court ruling on the BRCA gene patents came within a month of Angelina Jolie's New York Times editorial titled “My Medical Choice.” In this piece, she chronicles her experience with BRCA1 genetic testing and her decision to undergo prophylactic bilateral mastectomy after testing positive for a mutation. Ms. Jolie's international celebrity propelled her widely publicized editorial and the impact on public awareness has been studied. This impact has been dubbed the “Angelina Jolie Effect” to refer to the increase in awareness of genetic testing, information seeking about testing and prevention, and interest in genetic testing that followed 27–29.
LYNCH SYNDROME (LS)In 1913, Dr. Aldred Warthin, a pathologist, reported on four families with striking histories of multi-generational cancer ascertained from a review of the medical records of patients treated at the University of Michigan 30. Among these families was that of Dr. Warthin's seamstress, who later died of endometrial cancer. Her family, forever known as “family G” was notable for the preponderance for endometrial, gastric, and intestinal cancers and has been studied extensively over the years 30–34. In 1966, two large families from Nebraska and Michigan, with a similar spectrum of cancers, were reported on by Dr. Henry Lynch 35.
Lynch et al proposed that an autosomal dominant inheritance was the most likely etiology for cancer in these families 35, this cancer condition, characterized by multiple generations of early onset colorectal cancer without polyposis. The term hereditary non-polyposis colorectal cancer emerged to distinguish these families with colorectal cancer, from those with a familial adenomatous polyposis phenotype 36–38. Additionally, the terms Lynch syndrome I and Lynch syndrome II were utilized to differentiate between families with colorectal cancer specific histories (LS I) and families with colon cancer, as well as extra-colonic cancers, especially endometrial (LS II) 39,40.
To identify families with a clinical diagnosis of LS, the International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer established criteria, known as Amsterdam I criteria, in 1991. Amsterdam I criteria requires the presence of at least three relatives with colorectal cancer; one of whom is a first-degree relative of the other with at least two successive generation affected with cancer, as well as one family members diagnosed under age 50 41. However, Amsterdam I criteria would not account for other LS-associated cancer and limited the ability to identify LS families. Amsterdam II criteria were formulate to allow for the inclusion of diagnoses of extra-colonic LS-associated cancers in families 42.
MSH243,44 was the first mismatch repair (MMR) gene linked to LS in 1993, followed the next year by MLH145,46 and PMS2 47 and lastly by the discovery of MSH6 53 in 1997. In 2009, deletions in EpCam, (also known as TACSTD1) located upstream of MSH2, were implicated in LS 48. Initial molecular studies of LS demonstrated that most families had mutations in MLH1 and MSH2. However, more widespread genetic testing has shown that mutations in MSH6 and PMS2 were underappreciated.
While estimates of cancer risk vary by gene, individuals with LS are at elevated risk for colorectal cancer, endometrial cancer, gastric cancer, ovarian cancer, as well as cancers of the small intestine, hepatobiliary and urinary tracts, brain, and skin. Early studies of families who met clinical criteria for LS demonstrated over an 80% lifetime risk of colorectal cancer 49. More recent studies of MLH1 and MSH2 mutation carriers have estimated that women have a 43-52% risk for colorectal cancer and men a 66-69% risk 50,51. The colorectal cancer risk associated with MSH6 has been shown to be 20% for women and 44% for men 52. PMS2 colorectal cancer risk is estimated to be 15-20% 53. Women with LS also have a 25-60% risk for endometrial cancer 49,54. Gastric cancer risk is 6-13% and ovarian cancer risk is as high as 11% 55.
Beyond the defined familial cancer phenotype, distinct pathologic features of LS tumors were identified. Colon cancers in individuals with LS are more frequently right-sided and have characteristic histological features, with a tendency to be mucinous and poorly differentiated, with high levels of tumor-infiltrating lymphocytes. A hallmark of these tumors is their microsatellite instability (MSI). Microsatellites are tandem repeats in DNA, usually one to five base pairs long, which are located throughout the genome. Microsatellite instability refers to an abnormal number of these repeats in a tumor, either a reduction or expansion, compared to the normal tissue. This association of MSI-instability with familial colon cancer was reported in 1993, in two publication in the same edition of Nature, noting that MSI instability was correlated with tumors in the proximal colon 56 and was overrepresented in tumors from familial cases, compared to sporadic colorectal cancer 57. Similar findings were replicated later that year in individuals with sporadic and familial endometrial cancers, demonstrating that MSI-instability was more common amongst the familial cases 58. While not all MSI instable colorectal and endometrial cancers are attributable to LS, the majority these tumors in individuals with LS will be instable, making MSI instability an important indicator of cancer etiology. Sporadic MSI-instable colorectal cancer can be attributable to acquired hypermethylation of the MLH1 gene.
In 1997, the Bethesda guidelines were developed and updated in 2004, to identify which colorectal cancer patients should have MSI testing of their tumors performed 59,60. These guidelines were defined as individual with:
- 1.
Colorectal cancer diagnosed under age 50,
- 2.
Multiple primary colorectal cancers or other LS-associated tumors,
- 3.
Colorectal cancer with MSI-high histology diagnosed under age 60,
- 4.
Colorectal cancer diagnosed in one or more first-degree relatives with a LS-associated cancer, one of which was diagnosed under age 50, or
- 5.
Colorectal cancer diagnosed in two or more first – or second–degree relatives with LS-associated tumors, regardless of age at diagnosis.
However, in 2008 Hampel et al. published on a series of unselected colorectal cancer patients who had both MSI and immunohistochemical staining (IHC) of the MMR proteins performed, with subsequent germline sequencing for those with abnormal tumor studies. This study found that 3.6% of colorectal cancer patients had LS, but 28% of these individuals would have been missed if tumor studies had been restricted to Bethesda guidelines 61. In 2009, the Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group found that there was sufficient evidence to recommended for the genetic evaluation for LS in all newly diagnosed colorectal cancer patients 62. Universal screening of colorectal and endometrial cancer patients, either by MSI or IHC of the MMR genes, has been variably successful in its implementation, but it is clear that utilizing Amsterdam criteria or Bethesda guidelines alone will miss a significant proportion of LS families. Thus, broader criteria, in conjunction with tumor testing are recommended strategies to identify families with LS 62–64.
Several studies published in 2014, have added to our understanding of the underlying etiology of mismatch repair deficiency in colorectal and endometrial cancers 65–67. These studies demonstrated that 52-69% of tumors with that have unexplained mismatch repair deficiency are attributable to multiple somatic MMR gene mutations or loss of heterozygosity (LOH). This is clinically significant advancement, as individuals with unexplained MMR deficient tumors, especially loss of MSH2 and MSH6 by IHC, were often managed as having LS, despite the lack of a detectable germline mutation.
CONCLUSIONHistorically, individuals at risk for the syndromes reviewed here, as well as other hereditary cancer syndromes were identified using targeted genetic testing of the gene(s) highest in the differential diagnosis formulated from the phenotype. Therefore, genetic testing was highly selective by nature, driven by our understanding of the expected phenotype. This is exemplified by the differences in early estimations of cancer risk in BRCA families, which came from the highly selected linkage families, compared to the lower estimates that have been generated from more broadly ascertained cohorts. Today, the increased utilization of broad multi-gene panel testing allows for the simultaneous assessment of various genes. This has increased the number of individuals identified to carry mutations in cancer predisposition genes 68–71. In addition, many panels also include genes that may not be part of those included in the differential. As such, there is a growing body of literature reporting on the identification of high-risk mutations in individuals with cancer diagnoses not traditionally associated with the germline mutation they are found to carry While these findings can lead to challenges with translating results to clinical management, our understanding of how we have defined syndromes in the past is likely to become more fluid in the future 72–74.
The author declare no conflicts of interest, in relation to this article.