





Desde la descripción inicial efectuada por Pick hace más de un siglo atrás, el interés por las demencias frontotemporales ha tenido un gran crecimiento. Gracias a los avances de las neuroimágenes y el desarrollo de biomarcadores, el progreso en este campo ha generado nuevos conocimientos en torno a la categorización clínica, correlatos neuronales de las funciones cerebrales superiores y mecanismos de neurodegeneración. Paradójicamente, estos avances han traído consigo cierta controversia y desafíos, los cuales han motivado la creación de nuevos criterios diagnósticos. Esta breve revisión muestra la evolución histórica de estas condiciones y describe los criterios diagnósticos actuales para la variante conductual y las afasias progresivas primarias, que son las dos formas clínicas de las demencias frontotemporales.
Since the seminal Pick's description more than a century ago, the interest in Frontotemporal dementias has grown exponentially. Fueled by advances in neuroimaging and biomarkers, progress in the field has brought new insights into clinical categorization, neural correlates of higher brain function and pathological mechanisms of neurodegeneration. Paradoxically, those advances have also brought controversy and challenges, some of which have motivated the inception of new diagnostic criteria for those conditions. This brief review accounts for the historical evolution of these conditions and describes current diagnostic criteria for behavioural and primary progressive aphasia, the two major clinical presentations of frontotemporal dementias.
Las demencias frontotemporales (DFT) tienen una prevalencia estimada de 15-22 casos/100.000 habitantes y son la causa más común de demencia en adultos menores de 65 años, siendo la edad de presentación más frecuente entre los 50-60 años 1. Afecta tanto a hombres como mujeres, aunque clásicamente la forma conductual ha sido descrita mayoritariamente en hombres.
Desde su primera descripción clínico-patológica hace más de un siglo, el interés por este tipo de demencia se ha incrementado notablemente, tal como se refleja en la última década por el número creciente de publicaciones en el tema. Este interés ha sido en gran parte generado por los avances en neuroimágenes y biomarcadores, el desarrollo de nuevas técnicas de análisis neuropatológico y no menos importante, una mejor caracterización clínica y emergencia de nuevos modelos neurocognitivos. Sin embargo, la plétora de información ha traído consigo cierta confusión entre investigadores y clínicos, en parte fomentada por el uso de múltiple terminologías y definiciones para referirse a una misma entidad y sobre todo, por el intento de conciliar información clínica con la proveniente de los estudios neuropatológicos. En este último ámbito, el descubrimiento de casos con enfermedad de Alzheimer (EA) que se presentan clínicamente con una variante frontal-conductual o, en otros casos, afásica, ha sido uno de los desafíos emergentes en el estudio de las demencias. Como respuesta a estos desafíos, investigadores clínicos de diversos centros internacionales han propuesto nuevos criterios para diagnosticar y clasificar este grupo de condiciones. Estos criterios buscan sentar una terminología en común, que trascienda diferencias locales, y que provean una base para rotular casos y compartir información clínica. Dada la relativa ausencia de información en nuestro medio, esta revisión expone el estado del arte en el diagnóstico de demencia frontotemporal desde el punto eminentemente clínico. En la primera parte, discutiremos brevemente la evolución conceptual de este grupo de demencias, lo cual es relevante para entender cómo surgen los criterios diagnósticos actuales, y luego nos referiremos a los criterios para las variantes conductuales y afásicas de demencia frontotemporal.
RESEÑA HISTÓRICAEl estudio contemporáneo de las demencias se remonta hace más de un siglo atrás cuando se establecieron las primeras asociaciones clínico-patológicas. La descripción detallada de cambios conductuales y cognitivos, junto con el desarrollo de nuevas tinciones histológicas permitió relacionar síndromes demenciales con cambios histopatológicos específicos. De este modo, Arnold Pick describió una serie de casos que presentaban cambios de personalidad y alteraciones conductuales acompañadas con un deterioro del lenguaje o afasia, y aún más relevante, atrofia cerebral focalizada en la parte anterior de los lóbulos temporales y frontales 2. Esta descripción contrastaba, sin embargo, con la idea predominante en esa época que sostenía que las demencias eran causadas por una atrofia cerebral difusa. Durante la primera mitad del siglo pasado, sucesivas series clínico patológicas 3–5 no solamente confirmaron la existencia de ‘demencias focales’ sino que también demostraron que ellas tenían cambios histopatológicos inespecíficos que ciertamente diferían de los descritos previamente por Alois Alzheimer 6. En esas series, sin embargo, se hizo hincapié a los cambios de personalidad y desajustes sociales, mientras que los déficits en otros dominios cognitivos, en particular lenguaje, no fueron considerados tan relevantes.
Durante y después de la segunda guerra mundial hubo un cierto abandono en el tema, hasta que su interés resurgió en la década de los ochentas con el advenimiento de las técnicas modernas de neuroimágenes. Estos avances posibilitó la identificación síndromes cognitivos progresivos que de otra manera hubiesen sido atribuidos accidentes vasculares u otras patologías cerebrales focales (Marsel Mesulam, comunicación personal). Es así como Mesulam presentó una serie de seis casos clínicos que presentaban afasia progresiva asociada a atrofia inespecífica de las regiones peri-Silvianas izquierdas sentando de este modo las bases para proponer una definición, aún ampliamente aceptada, de afasia progresiva primaria (APP). Afasia progresiva primaria engloba a un grupo de procesos neurodegenerativos que causan un deterioro insidioso del lenguaje, pero con relativo respeto de otros dominios cognitivos. Mesulam estableció un periodo empírico de al menos dos años de evolución para así descartar otras etiologías como enfermedades por priones 7.
Los avances en patología molecular, por otra parte, demostraron la existencia de numerosos marcadores moleculares específicos que diferían de los cambios histopatológicos propios de la enfermedad de Alzheimer y causaban atrofia en la parte anterior de los lóbulos temporales y frontales. A este grupo de condiciones se les denominó colectivamente degeneración lobar frontotemporal, y se asociaron a diversos síndromes neurocognitivos. De paso, esta denominación aún causa confusión, ya que aunque es ampliamente usada por patólogos, en círculos clínicos es más habitual referirse simplemente como demencia frontotemporal. De cualquier modo, las correlaciones clínica patológicas modernas, evocando las primeras descripciones, no solamente enfatizaron cambios conductuales en estos pacientes, sino que también demostraron la relevancia de la presencia de signos frontal y disfunción executiva propias de lesiones pre-frontales.
Esto motivó la primera formalización de criterios clínico-patológicas 8 los cuales se basaron en el reporte retrospectivo de casos con confirmación patológica. Sin embargo, también fue evidente que hasta la mitad de los pacientes presentaban alteraciones del lenguaje, muchos de los cuales coincidían con el perfil neuropsicológico previamente descrito por Elizabeth Warrington 9. Estos pacientes, aunque mostraban un lenguaje fluido, presentaban defectos notorios en la comprensión de palabras e identificación de personas u objetos, alteraciones todas que son atribuidas a una pérdida gradual de la memoria semántica. De manera muy interesante, estos casos preservaban ambos hipocampos, los cuales típicamente se afectan en la EA, pero mostraban una conspicua atrofia de ambos polos temporales, a menudo lateralizada a izquierda 10. Sucesivas descripciones también reportaron casos con afasias no-fluentes, caracterizada por defectos en la producción del lenguaje que abarcaban alteraciones en el habla y reducción general de la capacidad de generar palabras y oraciones sintácticamente correctas 11. Era claro que la diversidad sindrómica no podía ser reducida a una sola forma clínica, sino que a varias presentaciones. Esto motivó la elaboración de nuevos criterios diagnósticos, en que las formas clínicas se separaron entre presentaciones conductuales o ‘frontales’ 12 y presentaciones afásicas -cuya definición en gran parte se homologó a las definiciones de APP acuñada por Mesulam-, y que a su vez de subdividieron entre afasias fluentes y no-fluentes.
SURGIMIENTO DE LOS CRITERIOS ACTUALESA pesar de la mejor delineación nosológica, los criterios mencionados presentaban algunas limitaciones. Muchas de las manifestaciones conductuales de la DFT variante conductual son difíciles de catalogar y objetivar mediante pruebas neuropsicológicas y en gran parte, su caracterización depende del relato de un familiar o un informante confiable. Adicionalmente, se demostró que una proporción de casos de variante conductual (DFTvc) progresaban de manera muy lenta, poniendo en duda la etiología neurodegenerativa. Por otra parte, otros casos presentaban cambios cerebrales evidentes a las neuroimágenes o portaban mutaciones genéticas que se habían establecidas como causales de estas demencias (ver abajo). Estas diferencias en la certeza diagnóstica motivaron la creación de nuevos criterios para la variante conductual 13, en los cuales los casos se consideraron tres categorías diagnósticas: posible, probable o definitivo (Tabla 1).
CONSENSO INTERNACIONAL DE CRITERIOS PARA DEMENCIA FRONTO-TEMPORAL VARIANTE CONDUCTUAL DFTVC
| I. Enfermedad neurodegenerativa |
|---|
| El siguiente síntoma debe estar presente para cumplir criterios de DFTvc. |
| A. Muestra deterioro progresivo de conducta y/o cognición por la observación o por la historia clínica aportada por un informante confiable. |
| II. Posible DFTvc |
| Tres de los siguientes síntomas cognitivo/conductuales (A-F) deben estar presentes para cumplir criterios. Los síntomas deben ser recurrentes o persistentes, no sólo un evento único o muy infrecuente. |
| A. Desinhibición temprana* del comportamiento (uno de los siguientes síntomas A1-A3 debe estar presente): |
| A.1 Conducta social inapropiada |
| A.2. Pérdida del decoro y modales |
| A.3. Impulsividad, acciones descuidadas |
| B. Apatía o inercia temprana (uno de los siguientes síntomas B1-B2 debe estar presente): |
| B.1 Apatía |
| B.2 Inercia |
| C. Pérdida temprana de la simpatía o empatía (uno de los siguientes síntomas C1-C2 debe estar presente): |
| C.1 Respuesta disminuida frente a necesidades o sentimientos de otras personas. |
| C.2. Disminución del interés social, relaciones interpersonales o calidez personal. |
| D. Conductas perseverativas tempranas, conductas estereotipadas compulsivas/ritualistas (uno de los siguientes síntomas D1-D3 debe estar presente): |
| D.1 Movimientos simples repetitivos |
| D.2 Conductas compulsivas o ritualistas complejas |
| D.3 Lenguaje estereotipado |
| E. Hiperoralidad y cambios dietarios (uno de los siguientes síntomas E1-E3 debe estar presente): |
| E.1 Cambios en las preferencias alimentarias (particularmente carbohidratos). |
| E.2 Ingerir alimentos compulsivamente, aumento del consumo de alcohol o cigarrillos. |
| E.3 Exploración oral o consumo de objetos no comestibles. |
| F. Perfil neuropsicológico: déficits ejecutivos con relativa preservación de funciones de memoria y visuoespaciales (todos los siguientes síntomas F1-F3 deben estar presentes): |
| F1. Déficits en tareas ejecutivas. |
| F2. Conservación relativa de la memoria episódica |
| F3. Conservación relativa de tareas visuoespaciales. |
| III. Probable DFTvc. Todos los siguientes síntomas (A-C) deben estar presentes para cumplir criterios |
| A. Cumple criterios para posible DFTvc |
| B. Presenta declinación funcional significativa (de acuerdo a reporte del cuidador, o evidenciado por cuestionario de actividades de la vida diaria (Ej: CDR). |
| C. Los estudios de imágenes deben ser consistentes con el diagnóstico de DFTvc (uno de los siguientes C1-C2, debe estar presente): |
| C.1. Atrofia frontal y/o temporal en RM o TC |
| C.2. Hipoperfusión o hipometabolismo frontal y/o temporal en PET o SPECT |
| V. Criterios de exclusión |
| Criterios A y B deben estar contestados negativamente para diagnóstico de DFTvc. Criterio C puede ser positivo para posible DFTvc pero debe ser negativo para probable. |
| A. El patrón de déficits es mejor explicado por otras enfermedades no neurodegenerativas o condiciones médicas. |
| B. La alteración conductual es mejor explicada por un diagnóstico psiquiátrico. |
| C. Los biomarcadores indican fuertemente el diagnóstico de enfermedad de Alzheimer u otra enfermedad neurodegenerativa. |
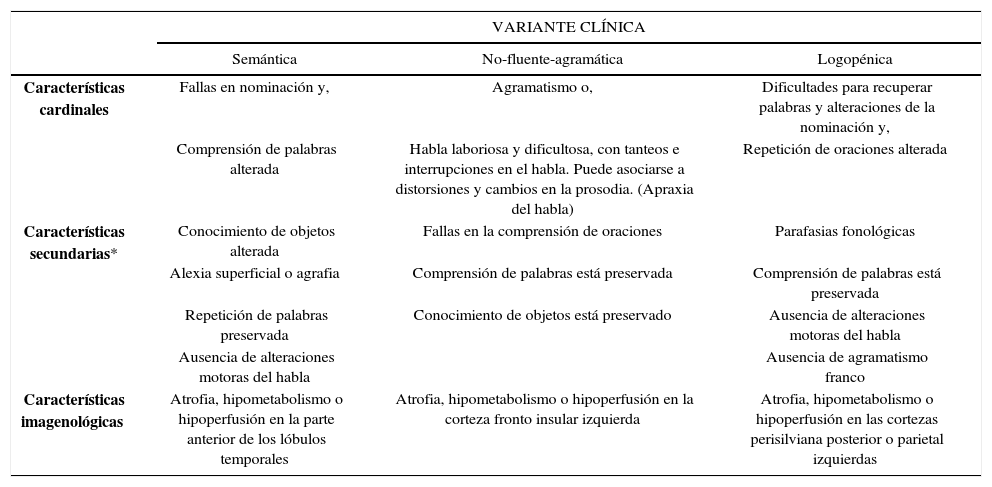
Por otra parte, numerosas series clínico-patológicas evidenciaban que hasta un tercio de los casos con APP eran causados por EA. Estos APP casos fueron luego identificados con un síndrome afásico distintivo: afasia logopénica 14. Esta tercera APP variante, junto con la no-fluente y la fluente (referida también como variante semántica), fueron formalmente definida por consenso como los criterios actuales (Tabla 2) 15.
Criterios para la clasificación de variantes clínicas de afasia progresiva primaria
| VARIANTE CLÍNICA | |||
|---|---|---|---|
| Semántica | No-fluente-agramática | Logopénica | |
| Características cardinales | Fallas en nominación y, | Agramatismo o, | Dificultades para recuperar palabras y alteraciones de la nominación y, |
| Comprensión de palabras alterada | Habla laboriosa y dificultosa, con tanteos e interrupciones en el habla. Puede asociarse a distorsiones y cambios en la prosodia. (Apraxia del habla) | Repetición de oraciones alterada | |
| Características secundarias* | Conocimiento de objetos alterada | Fallas en la comprensión de oraciones | Parafasias fonológicas |
| Alexia superficial o agrafia | Comprensión de palabras está preservada | Comprensión de palabras está preservada | |
| Repetición de palabras preservada | Conocimiento de objetos está preservado | Ausencia de alteraciones motoras del habla | |
| Ausencia de alteraciones motoras del habla | Ausencia de agramatismo franco | ||
| Características imagenológicas | Atrofia, hipometabolismo o hipoperfusión en la parte anterior de los lóbulos temporales | Atrofia, hipometabolismo o hipoperfusión en la corteza fronto insular izquierda | Atrofia, hipometabolismo o hipoperfusión en las cortezas perisilviana posterior o parietal izquierdas |
Adaptado de Gorno-Tempini et al (2011).
*Al menos 3 de 4 características secundarias deben estar presente en la variante semántica y variante logopénica y 2 de 3 en la variante no-fluente-agramátical.
En los últimos años ha existido un creciente desarrollo en torno a la genética de la DFT. Sin embargo, a pesar de estos avances, la relación exacta entre los genes identificados y la patología encontrada todavía no está del todo aclarada.
Hasta ahora se reconoce cerca del 40-50% de los pacientes puede tener historia familiar no bien definida y que 10-15% de los casos están asociados con una mutación genética autosómica dominante.
Las variaciones genéticas más frecuentes incluyen mutaciones del gen MAPT (microtubule associated protein tau, cromosoma 17), mutaciones en el gen GRN (progranulin, cromosoma 17) y la más reciente descrita, la expansión del hexanucleótido GGGCCC del gen c9orf72 (cromosoma 9), que da cuenta de familias presentando el fenotipo de DFT y también de esclerosis lateral amiotrófica (ELA) 16,17.
También existe asociación a otras mutaciones genéticas, menos frecuentes, incluyendo el gen CHMP2B, gen VCP (Valosin containing protein), gen SQSTM1 (sequestosome 1) y UBQLN2 (ubiquiline 1). Las variantes en los genes TARDBP (Tar DNA binding protein) y FUS (fused sarcoma) parecen ser más frecuentes en los casos asociados a DFT-ELA.
Recientes estudios utilizando la técnica GWAS (Genome wide association study) han revelado la asociación de DFT con TMEM106B (modifying factor transmembrane protein 106B), otros dos loci RAB38 (member RAS oncogene family) y CTSC (catepsin C) y otro relacionado a un locus HLA.
ANATOMÍA PATOLÓGICAMacroscopíala DFT se caracteriza por una marcada atrofia en regiones frontotemporales, que puede llegar a ser tan severa, produciendo que los giros adquieran un aspecto descrito como en ‘filo de navaja o cuchillo’.
Los hallazgos neuropatológicos clásicos son gliosis cortical y subcortical, pérdida cortical de neuronas y microvacuolación.
MicroscopíaEl estudio inmunohistoquímico de la degeneración lobar frontotemporal revela varios patrones de acuerdo a las inclusiones proteicas que se encuentren depositadas 18,19.
El primer patrón es identificado por la acumulación de proteína TAU, en la forma clásica de cuerpos de Pick o como depósitos difusos en neuronas y glía. Esta forma es encontrada en casos con mutación del gen MAPT. Dentro de los subtipos clínicos, representa casi el 45% de los casos de DFTvc y la mayoría de los casos de APP no fluente/agramatical (APPnfv). Así también la parálisis supranuclear progresiva (PSP) 20 y la degeneración corticobasal (DCB), presentan depósitos de proteína TAU en la anatomía patológica.
El segundo tipo está asociado a depósito de ubiquitinas. El principal subtipo, es la TDP-43 (Transactive response binding DNA protein, 43kDa), que participa en un amplio espectro de procesos ligando ADN, ARN y también otras proteínas. Suele ser una proteína nuclear neuronal y glial, pero en su estado patológico disminuye su concentración normal en núcleo, para conformar depósitos intranucleares o citoplasmáticos. En la nomenclatura este subtipo patológico recibe el nombre de FTLD-TDP. Esta variante se encuentra en mutaciones del gen TARDBP, pero también en mutaciones de GRN y repetición de exón en c9orf72. Dentro del espectro clínico, representa casi el otro 50% de los casos de DFTvc y la mayoría de los casos con APP semántica. Es destacable también que esta forma de depósito es la más frecuentemente encontrada en la esclerosis lateral amiotrófica y en aquellos casos de asociación entre DFT-ELA.
Otro tipo de inclusión, también ubiquitin positiva, es la de FUS/TLS (fused in sarcoma/translated into liposarcoma, de 526 aminoácidos), asociada a mutación del gen FUS. Es también una proteína ligadora de ADN y ARN, pero es menos frecuente, representando un 5% de los casos.
FORMAS CLÍNICASI. DEMENCIA FRONTOTEMPORAL VARIANTE CONDUCTUAL
Manifestaciones clínicasEste tipo de presentación se caracteriza por un cambio evidente en la personalidad y conducta del paciente, respecto a su comportamiento habitual.
Frecuentemente se presenta apatía, que el paciente puede manifestar por un menor interés en sus asuntos laborales, sociales, recreacionales y familiares. Falta de iniciativa para tomar decisiones o realizar acciones, con aplanamiento emocional frente a situaciones personales o familiares estresantes.
Otro síntoma predominante es la desinhibición, que puede tener distintas manifestaciones. Los paciente pueden comenzar a presentar pérdida del tacto social, realizando una crítica abierta hacia otras personas, comentarios inapropiados de contenido sexual, pérdida de límites sociales y físicos (con aproximación a extraños o conocidos en contexto inadecuado). Presentan a menudo impulsividad, que puede derivar en discusiones con familiares o colegas, accidentes de tránsito, gastos excesivos y no justificados de dinero e incluso abuso de alcohol o drogas.
Es muy frecuente que desde el principio el paciente muestre pérdida de empatía, que asociada a su falta de conciencia de enfermedad, cause severos disturbios en el entorno familiar y social.
Al mismo tiempo, los pacientes pueden presentar conductas estererotipadas simples o ritualistas y estereotipias verbales 21,22.
Cambios en hábitos alimentarios suelen ser frecuentes, con hiperfagia y preferencia por alimentos dulces o por un tipo fijo de alimento 23.
Perfil neuropsicológicoInicialmente los pacientes con DFTvc pueden presentar un rendimiento normal o casi normal en pruebas de tamizaje como el MMSE.
En general pruebas de tamizaje más detalladas como Montreal Cognitive assessment (MOCA) Addenbroke¿s Cognitive Examination revised (ACE-R) pueden detectar déficits iniciales en estos pacientes. De hecho, el ACE-R ha demostrado detectar déficits en el 90% de los casos con DFTvc 24.
En una evaluación neuropsicológica lo que suele encontrarse es el déficit en funciones ejecutivas; principalmente en atención, memoria de trabajo, fluencia lexical y resolución de problemas. Recientes estudios han demostrado que los pacientes con DFTvc pueden tener fallos específicos en las pruebas de control inhibitorio del Hayling test, series de dígitos inversa, fluencia lexical y Trails parte B (TMT-B) 21,25.
Los pacientes con DFTvc también presentan déficits en cognición social. Pruebas de teoría de la mente como el Faux Pas, la habilidad para comprender sarcasmo y para reconocer emociones, pueden estar consistentemente alteradas incluso en etapas tempranas de la enfermedad 26–28. Aunque se plantea un menor compromiso de memoria episódica que en EA, esta afirmación es aun controversial 29,30. Sin embargo, clásicamente se espera que este tipo de déficit sea más prominente en EA.
NeuroimagenEl uso de RM estructural muestra atrofia frontal mesial, orbitofrontal y de corteza insular anterior, que pueden ser más evidentes en cortes coronales. En algunos casos es posible encontrar una combinación de atrofia frontal y temporal anterior bilateral asociado a atrofia de núcleos basales (Figura 1 A).
, APPvs (B), APPvnf (C) y APPvl (D). Nótese que DFTvc (A) muestra atrofia simétrica y marcada de los lóbulos frontales que se extiende a la parte anterior de los lóbulos temporales. La imagen de APPvs (B) muestra una atrofia evidente, asimétrica, de la parte inferior y anterior del lóbulo temporal, con cierto respeto del hipocampo. El patrón de atrofia en APPvnf, en cambio, muestra aumento del tamaño del valle silviano izquierdo a expensas de la atrofia de la ínsula, y giro frontal inferior. En la Figura D, se observa un caso con APPvl que también muestra expansión del valle silviano izquierdo, pero además atrofia en la región temporal superior.")
IMÁGENES DE RESONANCIA MAGNÉTICA NUCLEAR REPRESENTATIVAS DE CORTES CORONALES A LA ALTURA DEL QUIASMA ÓPTICO
Se muestran ejemplos representativos de cortes coronales a la altura del quiasma óptico que evidencian el patrón de atrofia cerebral para DFTvc (A), APPvs (B), APPvnf (C) y APPvl (D). Nótese que DFTvc (A) muestra atrofia simétrica y marcada de los lóbulos frontales que se extiende a la parte anterior de los lóbulos temporales. La imagen de APPvs (B) muestra una atrofia evidente, asimétrica, de la parte inferior y anterior del lóbulo temporal, con cierto respeto del hipocampo. El patrón de atrofia en APPvnf, en cambio, muestra aumento del tamaño del valle silviano izquierdo a expensas de la atrofia de la ínsula, y giro frontal inferior. En la Figura D, se observa un caso con APPvl que también muestra expansión del valle silviano izquierdo, pero además atrofia en la región temporal superior.
Sin embargo, debe tenerse en cuenta, que un estudio con RM cerebral normal no excluye necesariamente el diagnóstico, sobre todo en etapas iniciales de la enfermedad.
El desarrollo de nuevas técnicas como VBM (voxel-based morphometry) y mapeo de grosor cortical ha sido útil para poner en evidencia atrofia selectiva de el cíngulo anterior y corteza frontal insular en etapas tempranas de la enfermedad, lo que lo distingue de las variantes de lenguaje y de EA.
Estudios explorando la conectividad de redes también han mostrado cambios significativos en conectividad entre las regiones más sensibles a la atrofia.
La atrofia también puede afectar otras áreas como amígdala, hipocampo, caudado, estriado, putamen, tálamo e hipotálamo.
Estudios de sustancia blanca usando DTI (difussion tensor imaging), han permitido determinar cambios en la microestructura de la sustancia blanca. Pacientes con DFTvc muestran una reducción selectiva en algunos tractos, como el fascículo longitudinal superior, fascículo uncinado, cíngulo, cuerpo calloso, particularmente en el lóbulo frontal.
Estudios de seguimiento por RM han permitido establecer que existe una atrofia significativa a largo del tiempo. La atrofia cerebral puede alcanzar un 8% anualmente.
Estudios funcionales como el SPECT (99mTc hexamethylpropyleneamine oxime single-photon emission computed tomography) o FDG-PET (18F-fluordeoxyglucose) han colaborado también al diagnóstico.
En el SPECT es posible ver hipoperfusión frontal, así como en el FDG-PET es posible observar hipometabolismo en regiones frontales.
Manejo de la demencia frontotemporalEs de vital importancia informar a la familia o persona al cuidado del paciente que el cambio conductual es parte de la enfermedad, ya que la falta de empatía y los problemas de cognición social, pueden dificultar significativamente las relaciones interpersonales.
Tal y como es requerido en otros tipos de demencia, las medidas de cuidado general son muy importantes para el manejo conductual del paciente.
Entre ellas pueden encontrarse:
- •
Manejo conductual y seguridad.
- •
Información al cuidador.
- •
Seguridad y manejo de vehículos.
- •
Decisiones familiares: manejo de patrimonio familiar. Jubilación anticipada. Institucionalización temprana.
- •
Supervisión continua.
- •
Ejercicio y movilidad.
- •
Terapia ocupacional, fonoaudiología.
- •
Cuidado de rutinas, evitar factores externos que dificultan el manejo.
- •
Medio seguro y confiable.
- •
Cuidado del cuidador.
Dentro del manejo farmacológico, no existe ningún tratamiento específico para la enfermedad, sin embargo algunos fármacos pueden facilitar el manejo de los síntomas.
AntidepresivosEstudios con fármacos serotoninérgicos ISRS, han demostrado que sertralina, paroxetine y fluvoxamina disminuirían la desinhibición, ansiedad, impulsividad, el trastorno alimentario y las conductas repetitivas (resultados basados en estudios observacionales pequeños y estudios randomizados con resultados controversiales).
El uso de trazodona en dosis bajas: reduciría la agitación y la agresión (estudio doble ciego randomizado, muestra pequeña, n=26) 31.
AntipsicóticosSi se requiere este tipo de medicamentos, se sugiere usar aquellos atípicos, con bajas dosis, tales como olanzapina y quetiapina, para manejo de agitación, delirio y reducción del stress del cuidador.
AnticolinesterásicosExisten resultados controversiales. Donepecilo empeoraría los síntomas 32.
MemantinaExisten dos estudios randomizados fallaron en demostrar eficacia 33.
Otros: valproato, lamotrigina, carbamacepina: controversiales.
II DEMENCIA FRONTOTEMPORAL VARIANTE AFÁSICA
En esta categoría se incluye casos con DFT en que las alteraciones del lenguaje son el motivo principal de consulta e impactan el quehacer diario de los pacientes. En término práctico, para definir clínicamente estos casos se utiliza la definición de APP, ya indicada, propuesta por Mesulam. Posteriormente, se debe clasificar los casos en una de las tres categorías propuestas por los criterios internaciones (Tabla 2). La variante semántica se caracteriza por presentar fluencia conservada, con un lenguaje espontáneo que puede incluso llegar ser verborreico debido al uso excesivo e innecesario de palabras, sin que ellas aporten al contenido del mensaje. Estos pacientes muestran serias dificultades para entender el significado de las palabras, y consecuentemente, capacidad de denominar o evocar palabras. Por este “olvido de palabras”, no infrecuentemente estos pacientes son confundidos con EA; sin embargo, a diferencia de éstos casos con la variante semántica tienes preservada la memoria usada cotidianamente. Desde el punto de vista diagnóstico, la variante semántica es relativamente fácil de detectar mediante pruebas neurosicológicas capaces de evaluar procesamiento semántico 34. Aparte de las tareas de denominación, comprensión de palabras y vocabulario, las pruebas de asociación semántica, como las “Pirámides y Palmas” son altamente específicas y sólidas para confirmar el diagnóstico. Finalmente, estos casos presentan de manera muy constante y característica atrofia (o hipometabolismo) de los lóbulos temporales anteriores que es más evidente en cortes coronales (Figura 1B).
Los casos con la variante no fluente/agramátical (APP vnf) presentan diversas alteraciones de la fluencia expresiva del lenguaje determinadas por errores articulatorios, alteraciones en la prosodia, simplificación extrema del lenguaje, por ejemplo, habla telegráfica debida o la pérdida de las relaciones sintácticas entre las palabras. Cada uno de estos defectos puede presentarse de un caso a otro en grado variable, a fenómeno que demuestra la multidimensionalidad de las alteraciones de la fluencia. Por lo tanto es muy importante observar y detectar cada uno de estos defectos en forma separada. Así, se debe observar la presencia de errores articulares que corresponden a la falta de precisión o claridad de los sonidos del habla en palabras o frases. Éstos pueden estar acompañados de alteraciones de la prosodia, los cuales incluyen cambios en el ritmo, entonación y transición fluida entre sílabas. Alteraciones de la prosodia pueden producir un habla “cibernética o robótica” y entonación plana. Además, la velocidad del habla es lenta, mientras que el acento y ritmo o separación de las sílabas de las palabras y las frases esta alterado. De hecho, se han descrito casos incipientes con APPvnf que presentan cambios aislados en el acento al hablar, siendo APPvnf parte del diagnóstico diferencial de síndrome del acento extranjero 35. Alternativamente, hay casos que no presentan ninguna alteración motora del habla, pero sin embargo muestran alteraciones de la fluencia debidas a omisiones de palabras tales como preposiciones, artículos, pronombres y verbos auxiliares que resultan en la simplificación excesiva del lenguaje y en última instancia, habla telegráfica y agramatismo. Tal como se muestra en la Figura 1C, estos casos muestran atrofia asimétrica afectando la parte izquierda anterior del valle Silviano, principalmente área de Broca e insula.
Finalmente, la tercera y más recientemente variante clínica identificada es la variante logopénica. Ésta se caracteriza por la gran dificultad para evocar palabras, lo que se evidencia por pausas o latencias marcadas al hablar, que van acompañadas de muletillas (Interjecciones, “mmm”, “aah”), todo lo cual causa vacilaciones al hablar 36. Estos pacientes presentan alteraciones de la denominación que está en un punto intermedio con las otras variantes: no tan severo como la APPvs, pero no tan leve como la APPvnf 37. En contraste con las otras APP variantes clínicas, estos casos presentan relativa preservación de la memoria semántica y ausencia de alteraciones motoras del habla. Sin embargo, se ha sugerido que las parafasias fonológicas, que se definen por la sustitución o adición de segmentos fonológicos bien articulados u omisión de fonemas o sílabas enteras, podrían ser altamente específicas para esta variante 38. Además de estas alteraciones, estos casos presentan de manera muy marcada dificultades para repetir oraciones.
En contraste con las otras variantes, la distribución de la atrofia cerebral abarca las regiones retro-silvianas izquierdas, especialmente el girus temporal superior posterior y la región parietal inferior, abarcando el girus angularis (Figura 1D).
CONCLUSIONESEsta revisión muestra cómo el estudio de las DFTs esta cobrando relevancia en el campo de neurología cognitiva y demencias. Por un lado, debido que estas demencias selectivamente destruyen redes neuronales que sustentan la cognición social, lenguaje y memoria semántica, su estudio representa un paradigma natural para entender cómo las funciones cognitivas superiores se organizan en el cerebro. En contraste con la EA, una alta proporción de casos con DFT tienen antecedentes familiares con demencia o ELA, lo que puede facilitar el entendimiento de la cadena patogénica en estas demencias. Paradójicamente, el estudio de DFT ha entregado valiosa información acerca de EA, debido a que un porcentaje no despreciable de estos casos presentan con manifestaciones atípicas que son prácticamente indistinguibles de las presentaciones de la DFT. Esperamos que este breve revisión contribuya a entender la complejidad de este grupo de demencias y así mejorar el diagnóstico de casos con DFT.
Los autores declaran no tener conflictos de interés, en relación a este artículo.