




Las demencias neurodegenerativas se caracterizan por la pérdida selectiva y progresiva de poblaciones neuronales vulnerables, lo que determina la presentación clínica de éstas. La misma población neuronal puede comprometerse en diferentes enfermedades. Dado que la presentación clínica refleja las neuronas vulnerables del proceso patológico subyacente, es claro que más de una enfermedad puede dar cuenta de un síndrome clínico determinado. Estas enfermedades comparten como mecanismo patogénico la acumulación de agregados proteicos, a menudo asociado con toxicidad y una falla del clearence por vías de degradación celular. Para el propósito de esta breve revisión, se presentan los aspectos neuropatológicos más relevantes de las demencias degenerativas, de acuerdo a la clasificación clínica y la patología molecular subyacente.
Neurodegenerative dementias are characterized by selective and progressive loss of specific populations of neurons, which determines the clinical presentation. The same neuronal populations can be affected in a number of different disorders. Given that the clinical presentation reflects the particular population of neurons that are targets of the disease process, it is clear that for any given clinical syndrome, more than one neurodegenerative disease can account for the clinical syndrome. Neurodegenerative dementias share a common pathological pathway and this is the accumulation of protein aggregates, often associated with toxicity and failure of clearance by cellular degradation pathways. The disorders are referred to as ⿿proteinopathies⿿. For the purpose of this brief review, the major neuropathological aspects are presented according the clinical syndrome and the molecular pathology.
Las Demencias Neurodegenerativas (DN) comparten varios factores que permiten diferenciarlas de otras:
-
Comprometen grupos neuronales vulnerables específicos, generalmente separados espacialmente, pero relacionados en función y las características clínicas están determinadas por las estructuras comprometidas.
-
Los cambios histopatológicos se caracterizan por pérdida de neurona de las regiones afectadas con reacción astrocítica y microglial variable. Se piensa que las neuronas mueren por procesos no necróticos sino más bien apoptóticos.
-
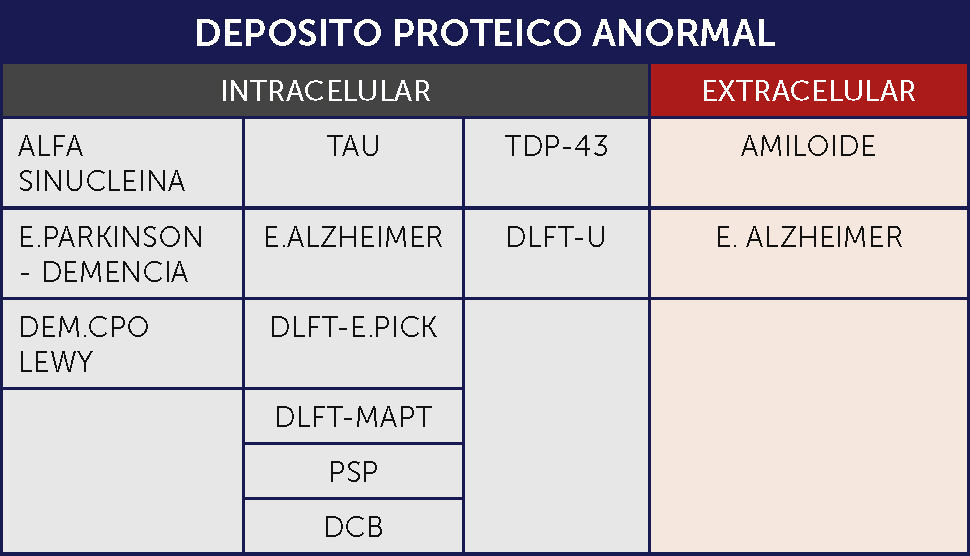
En muchas de las DN se acumulan proteínas específicas. Estas acumulaciones pueden ser intracelulares en neuronas o glía, originando inclusiones distintivas, tales como los ovillos neurofibrilares (ONF) o los cuerpos de Lewy o pueden ser extracelulares, formando ⿿placas⿿, tales como las placas neuríticas de la enfermedad de Alzheimer (EA).
Otrora condiciones enigmáticas y con poca comprensión de su patogénesis, la combinación de la biología celular para comprender los procesos que comparten y de la genética, han derivado en una reorganización de los esquemas de clasificación de algunas de estas enfermedades. Un hilo común de las DN es la acumulación de agregados protéicos, a menudo asociado con toxicidad y una falla del clearence por vías de degradación celular. Estos agregados son también evidentes en el examen histológico y por esta razón se refiere a estas enfermedades como proteinopatías. Para algunas se han descrito anormalidades genéticas bien caracterizadas, en otras se han visto casos asociados a mutaciones o bien, que pueden ocurrir ambas formas, esporádica y hereditaria 1⿿3.
CLASIFICACIÿNExisten varias aproximaciones a una clasificación de las DN, con diferentes ventajas cada una de ellas. De acuerdo al síndrome dominante de presentación clínica inicial, se han clasificado fundamentalmente en aquellas en las que predomina el deterioro cognitivo versus aquellas con trastornos motores, teniendo en consideración que el trastorno cognitivo siempre estará presente en cualquier fase de la evolución de la progresión de la enfermedad. Es esencial enfatizar, que con la progresión de la enfermedad, hay mucha sobreposición de la sintomatología. La clasificación basada en los sistemas anatómicos comprometidos se ha remplazado por una clasificación molecular-patológica (Figura 1) 1⿿3.
.")
Cuando se agrupan estas enfermedades en base a la acumulación proteica característica, las categorías incluyen:
Tauopatías: Enfermedades caracterizadas por la acumulación de la proteína asociada al microtúbulo TAU de acoplamiento alternativo (codificada por el gen MAPT). Tau es una fosfoproteína resistente al calor que promueve la polimerización y estabilización del microtúbulo. Las tauopatías primarias son aquellas enfermedades en las que se considera la patología Tau como el principal factor contribuyente a la neurodegeneración. Aunque inicialmente se consideraba restringida a las neuronas, actualmente se sabe que se acumula también en la glía en un amplio rango de enfermedades neurodegenerativas y en el cerebro envejecido. En las enfermedades neurodegenerativas la proteína Tau tiene una conformación anormal y propiedades de solubilidad anormal, que promueven la agregación y formación de fibrillas, similares al amiloide, excepto porque no se encuentran en el espacio extracelular, sino en el citoplasma de las células afectadas. En alguna de estas enfermedades hay mutaciones en el gen TAU incluyendo mutaciones puntuales así como mutaciones que alteran el acoplamiento. Las isoformas TAU pueden contener ya sea tres (3R) o cuatro (4R) de los dominios de unión del microtúbulo. Dentro de las tauopatías hay enfermedades caracterizadas por inclusiones con 3RTau (enfermedad de Pick), 4RTau (parálisis supranuclear progresiva-PSP-, degeneración corticobasal -DCB-) y ambos 3R y 4R (EA, en combinación con depósitos de abeta amiloide). En las variadas formas de DLFT puede haber 3R, 4R o una combinación de ambas 1⿿3.
Alfa Sinucleopatías: Enfermedades en las que hay acumulación de la vesícula sináptica asociada a la proteína Alfa Sinucleína. Pueden formarse agregados visibles en secciones de rutina con hematoxilina-eosina, o la acumulación puede ser sólo visible bajo tinciones especiales de inmunohistoquímica. Este grupo incluye la enfermedad de Parkinson (EP), la enfermedad por cuerpo de Lewy y la atrofia multisistémica (AMS) 1⿿3.
Proteinopatías TDP-43 (TDP-43): La TDP-43 es una proteína que se une al RNA/DNA implicada en el acoplamiento alternativo, regulación transcriptacional, estabilización del mRNA y procesamiento del micro RNA. Normalmente está presente el largo completo de la proteína en el núcleo. En condiciones patológicas, la proteína, fragmentada, fosforilada y ubiquitinada se acumula en el citoplasma del cuerpo celular, principalmente en neuronas, pero también en la glía. También se puede encontrar en neuritas. El interés en el TDP-43 surgió cuando se vio que era el componente de las inclusiones neuronales de la esclerosis lateral amiotrófica (ELA) y de la degeneración lobar fronto temporal con inclusiones ubiquitinadas (DLFT-U) 1⿿3.
PATOLOGÿA DE LAS DEMENCIAS DEGENERATIVAS QUE AFECTAN PRIMARIAMENTE LA CORTEZA CEREBRALENFERMEDAD DE ALZHEIMER (EA)La EA es la más frecuente de las DN y su incidencia se incrementa con la edad. La mayoría de los pacientes presenta trastornos de memoria y desarrolla déficits en otros dominios cognitivos, comúnmente incluyendo apraxia, afasia y agnosia.
Macroscópicamente es llamativa la atrofia de la corteza, la que compromete predominantemente el hipocampo, giro parahipocampal y la amígdala temporal, estando los lóbulos parietal y frontal menos severamente afectados. Habitualmente el lóbulo occipital se respeta. Hay dilatación ventricular de grado moderado, de acuerdo al grado de atrofia cortical. También es un hallazgo típico la despigmentación del locus coeruleus, con una preservación de la pigmentación de la sustancia nigra.
Microscópicamente las placas seniles y los ovillos neurofibrilares (ONF) han sido las lesiones características de la EA. La pérdida sináptica también es temprana y constante. Actualmente el diagnóstico neuropatológico de EA se basa en la combinación de depósitos de amiloide-beta (A-beta), de placas seniles y ONF, todos con la apropiada densidad y distribución 1. Los criterios de diagnóstico clínico de la EA han cambiado y actualmente descansan menos en la demostración de demencia severa, sino en vez consideran los estadíos más precoces de la enfermedad, cuando el deterioro cognitivo es menor. De la misma forma ha sido necesario desviar los criterios neuropatológicos para identificar el grado y distribución de las lesiones asociadas al proceso patológico subyacente pero que no están típicamente asociadas con el estado terminal del impedimento neurológico. 1,3⿿5.
Depósitos A-beta:El péptido A-beta es un fragmento de 40-42 amino ácidos de una proteína neuronal normal llamada APP por su sigla en inglés (proteína precursora del amiloide). Este péptido se genera por la división secuencial del APP en el dominio extracelular (por una proteasa conocida como BACE), seguido por un clivaje intramembranoso (por un complejo enzimático conocido como gama-secretasa, que incluye la presenilina). La generación del A-beta a partir del APP se considera crucial para el inicio de la patogénesis de la EA, dado que mutaciones del APP que aceleran la generación de A-beta causan las formas familiares de EA, así como lo hacen las mutaciones de la presenilina, las que incrementan la tasa de generación de A-beta o la desvían hacia formas más largas de A-beta. El descubrimiento reciente que una mutación del APP que retrasa la proliferación de A-beta es protectora contra la EA ha fortalecido aún más este argumento.
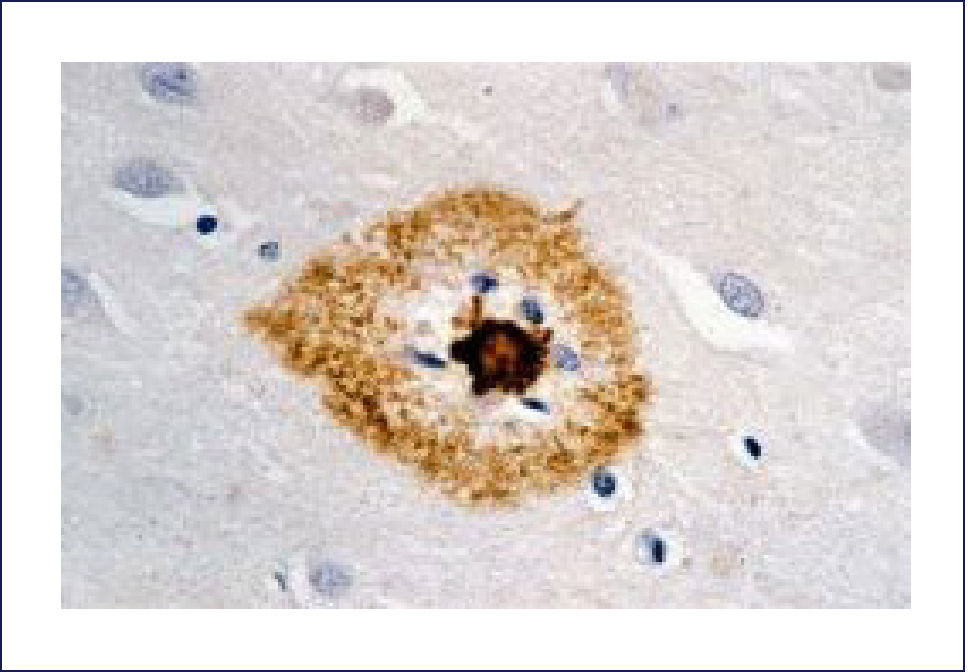
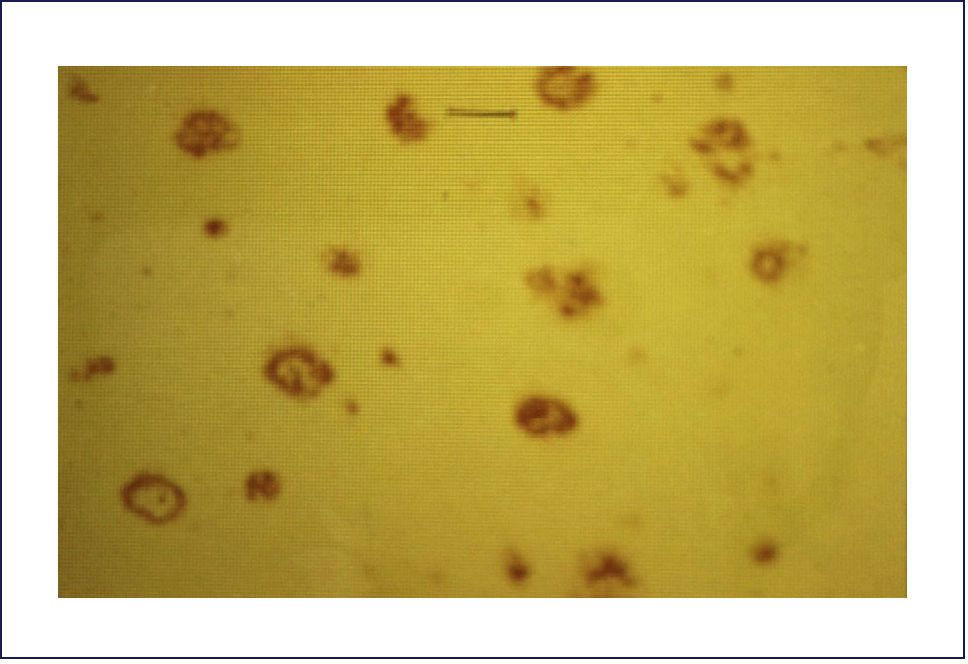
El A-beta, que se deposita extracelularmente, es muy susceptible de formar diferentes tipos de agregados, lo que morfológicamente se traduce en diferentes tipos de depósitos o placas; los pequeños agregados (llamados oligómeros) son actualmente los mediadores críticos sospechosos de desencadenar la disfunción celular y sináptica. El A-beta depositado comúnmente desencadena una reacción local significativa, lo que pasa a constituir la placa neurítica (Figura 2). Por otra parte se encuentran los depósitos A-beta difusos, de aspecto convulocionado y que no aparecen con las tinciones de rutina, pero son inmunoreactivos con Ac anti A-beta (Figura 3). La progresión de la patología amiloídea al interior del encéfalo (llamada progresión Thal) típicamente sigue una secuencia: 1, isocorteza, 2, hipocampo, 3, ganglios basales, 4 mesencéfalo, 5, protuberancia y cerebelo 1,3,6.
 lleva a la destrucción de la célula. (Foto: Hospital Universitario de Tübingen/ Mathias Jucker).")
.")
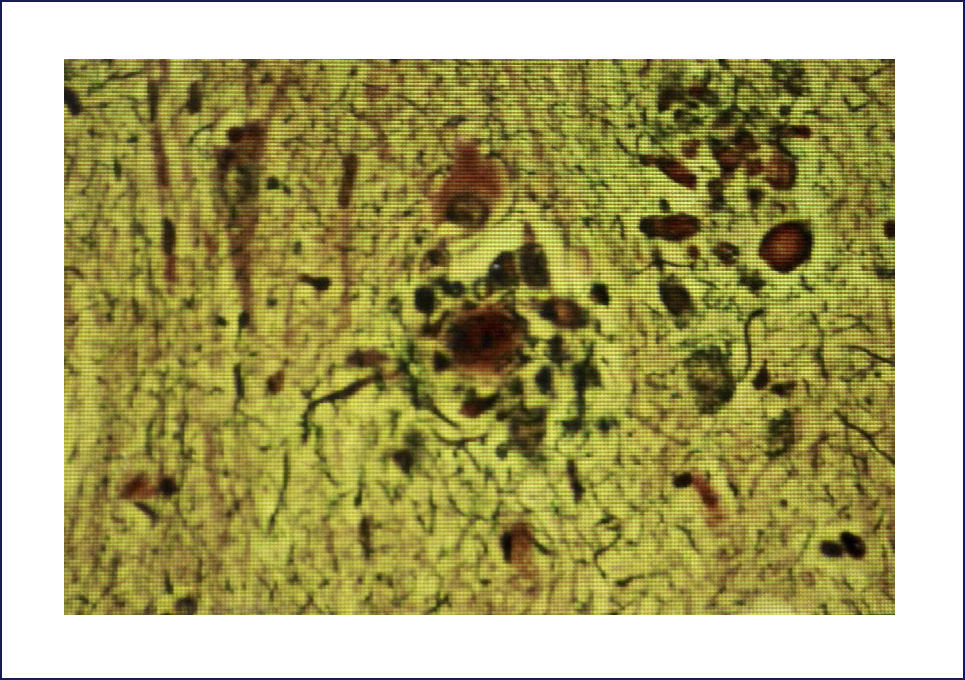
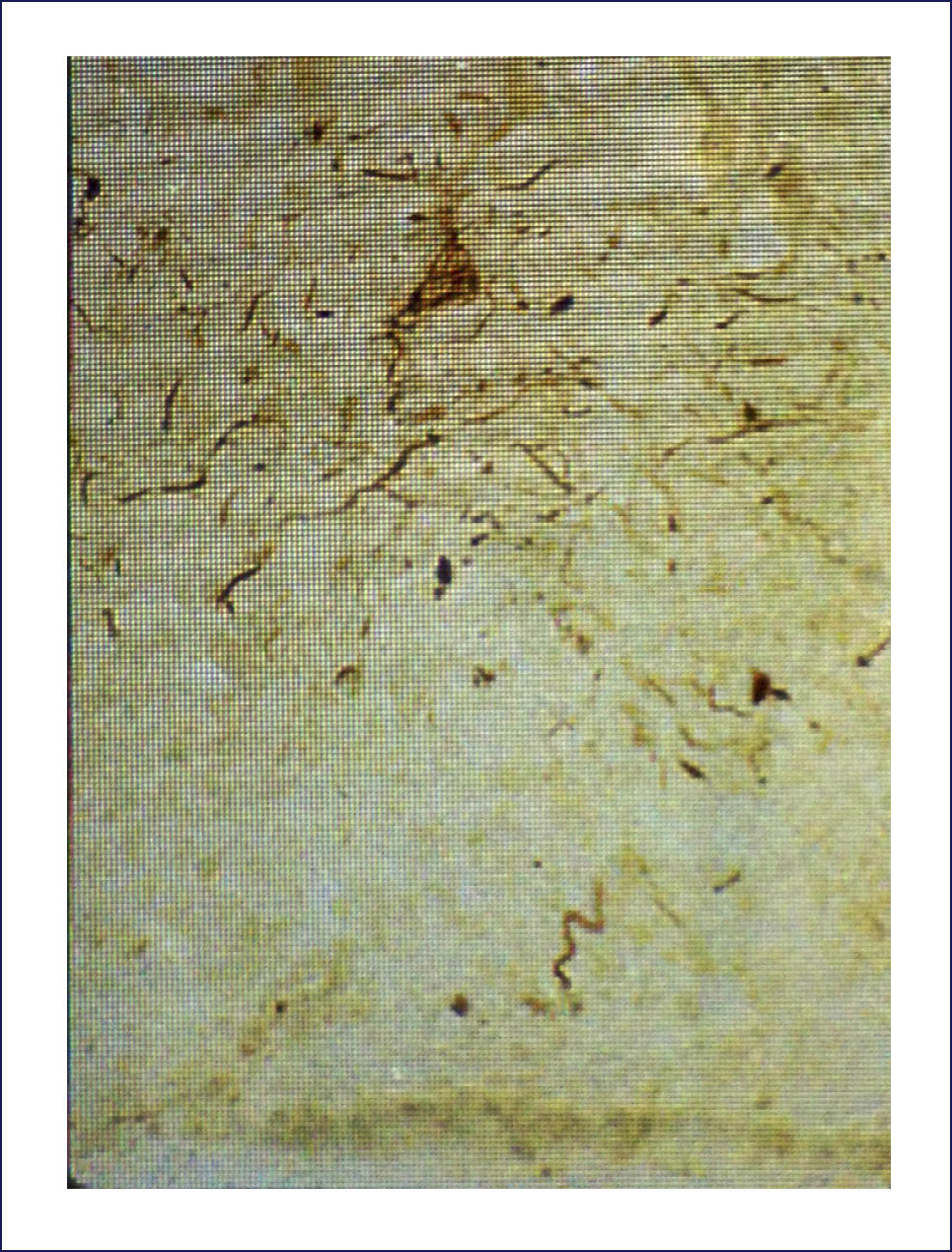
Las placas están formadas por un centro y una corona. El centro es un depósito extracelular de A-beta, mientras que la corona son neuritas distróficas, fundamentalmente axones, inmunoreactivos para TAU. El A-beta constituyente del centro de la placa senil está plegado y enriquecido de capas de beta y tiene toda las características de amiloide (muestra refringencia verde-manzana con la tinción de rojo congo, es fluorescente con tioflavina S) y tiene apariencia fibrilar en la Médula Espinal (ME). La placa neurítica desencadena una reacción astrocítica y microglial (Figura 4). Las placas neuríticas y los depósitos difusos se encuentran típicamente en la neocorteza, corteza entorrinal e hipocampo, mientras que el estriado y el cerebelo sólo tienen depósitos difusos 1. La intensidad de la patología de PN en la neocorteza se informa de acuerdo al sistema semicuantitativo CERAD en, escasas, moderadas y abundantes 7.
Ovillos neurofibrilares (ONF), pérdida neuronal y sináptica: rodeado por una corona de neuritas distróficas. (Foto: colección personal).")
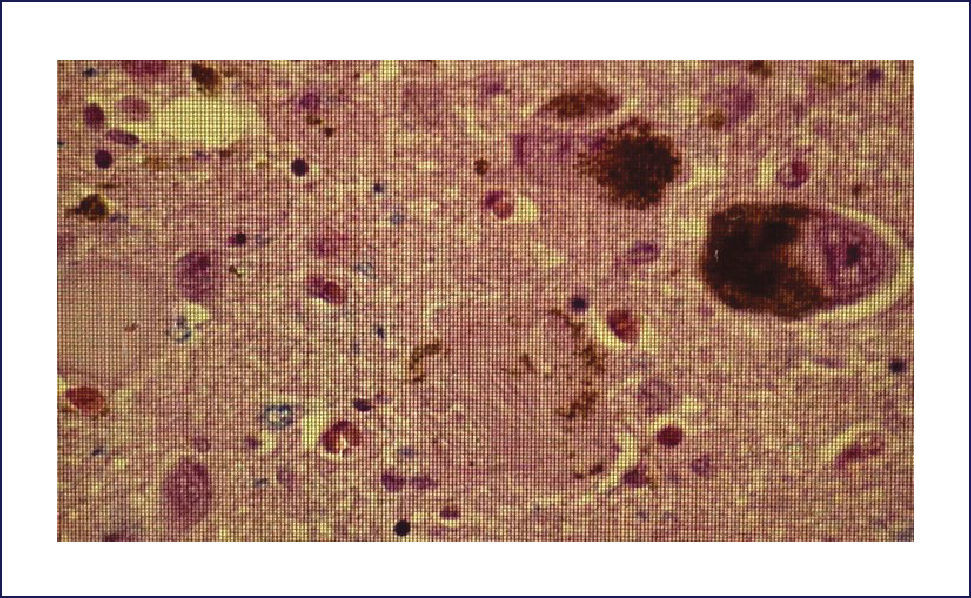
Los ONF son cuerpos de inclusión intracelulares, que principalmente contienen TAU y también otras proteínas. TAU es una proteína asociada a los microtúbulos, pero en el ONF está separada de estos y está hiperfosforilada. Los ONF se pueden detectar con inmunohistoquímica usando Ac para la proteína TAU, así también con técnicas de impregnación de plata y otras (Figura 5). Cuando la célula muere, los ONF quedan en la neuropila como ⿿ovillos fantasma⿿. Los procesos neuronales, especialmente dendritas, pueden también acumular TAU, tomando el nombre de neuritis distróficas. Tanto los ONF como las neuritis distróficas contienen las formas 3R y 4R de TAU 1.
.")
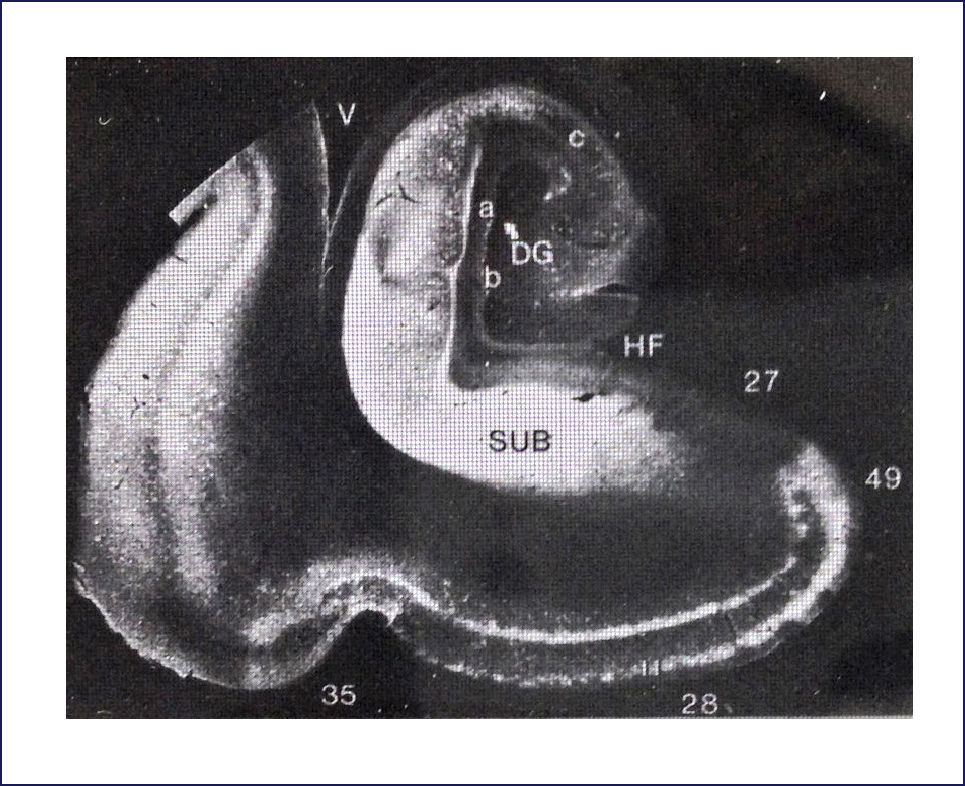
El esquema llamado los Estadíos de Braak y Braak se utiliza para caracterizar la distribución de la patología neurofibrilar, con una buena correlación con el estado clínico de la enfermedad. En general, los ONF y las neuritis distróficas aparecen primero en la corteza entorrinal (Estadío I), progresan a través del subículo (Estadío II) hacia el hipocampo, primero a CA1 (Estadío III) y así hacia el resto de las células piramidales (Estadío IV), antes de alcanzar cortezas de asociación (Estadío V) y finalmente se extienden hacia la neocorteza motora y sensitiva primarias (Estadío VI) (Figura 6). Además los ONF se encuentran en otras regiones, tales como amígdala, núcleos límbicos talámicos, etc. 8,9.
Criterios diagnósticos y Estadío de la EA. (Foto: colección personal).")
Se cuenta con tres escalas por las cuales se evalúan las lesiones en la EA: los ONF se tipifican de acuerdo a los estadíos de Braak para EA, los depósitos de Abeta de acuerdo a los evadíos de Thal y las placas neuróticas de acuerdo al sistema de CERAD 6⿿8.
De acuerdo al consenso de expertos más reciente, por definición los cambios neuropatológicos de la EA necesariamente incluyen depósitos Abeta, con cualquier combinación de PS y ONF 5. La progresión y el incremento de la patología de acuerdo a los estadíos descritos están asociados con una mayor probabilidad de daño cognitivo de los individuos afectados. En un extremo están los sujetos presumiblemente cognitivamente intactos, en una fase 1 de Thal de depósitos Abeta, con bajo estadío de Braak y escasas PN de acuerdo al CERAD, y al otro extremo hay casos con extensos depósitos Abeta (fase 5 de Thal), abundantes PN y ONF por toda la neocorteza y lóbulos temporales mesiales (Braak V o VI) 5.
DEGENERACIÿN LOBAR FRONTOTEMPORAL (DLFT)La DLFT es un término genérico para el grupo de demencias degenerativas no-Alzheimer las que se caracterizan por neurodegeneración concentrada en los lóbulos frontales y temporales con preservación relativa de los lóbulos parietales y occipitales. Clínicamente se caracterizan por trastorno conductual o disfunción del lenguaje, siendo el trastorno de memoria de aparición más tardía. La condición clínica se llama demencia fronto-temporal (DFT) y el sustrato patológico es la degeneración lobar fronto-temporal (DLFT) 1⿿3,10.
Los hallazgos histopatológicos, así como las bases genéticas que actualmente se conocen, han refinado la clasificación de este grupo de enfermedades. Actualmente se caracterizan de acuerdo al tipo de inclusión proteica encontrada en las neuronas, así como el estatus de mutaciones, si es conocido 10. Con el progreso de la investigación en este campo, la clasificación de estas enfermedades ha estado en un continuo proceso de actualización y actualmente se basa en el reconocimiento de alguno(s) de estos tres marcadores demostrados por la inmunohistoquímica: TAU, en varias combinaciones de 3R/4R (DFLT-TAU); TDP-43, una proteína que une DNA/RNA (DLFT-TDP-43); y FUS (DLFT-FUS). Dentro de cada uno de estos grupos, el patrón de distribución de las inclusiones y los marcadores genéticos son heterogéneos 10. Del punto de vista clínico los síntomas característicos permiten formar grupos de DLFT; estos reflejan la distribución anatómica de la pérdida neuronal más que el tipo de inclusión. Entre las principales formas de presentación, las tres mejor caracterizadas son la variante conductual de DLFT (vcDLFT), la afasia no-fluente primaria progresiva (APP) y la Demencia Semántica. Es importante destacar la falta de correlación entre la presentación clínica y los hallazgos patológicos; por ejemplo, en un estudio bien caracterizado de sujetos con APP se encontró en la autopsia que aproximadamente la mitad correspondía a EA y la mitad tenía una forma de DLFT, la que se dividía más o menos igual entre DLFT-tau y DLFT-TDP-43 1,10.
DLFT-tauEstas enfermedades se definen por la combinación de degeneración lobar e inclusiones que contienen TAU 3R, 4R o ambas formas. Este grupo incluye la enfermedad de Pick, las Tauopatías con mutación del gen TAU (MAPT) y otras Tauopatías sin mutación. Adicionalmente, otras dos enfermedades que se clasifican primariamente como Enfermedades de Movimientos Anormales (PSP y DCB) también pueden tener trastorno cognitivo con atrofia lobar e inclusiones que contienen TAU 1,2,10.
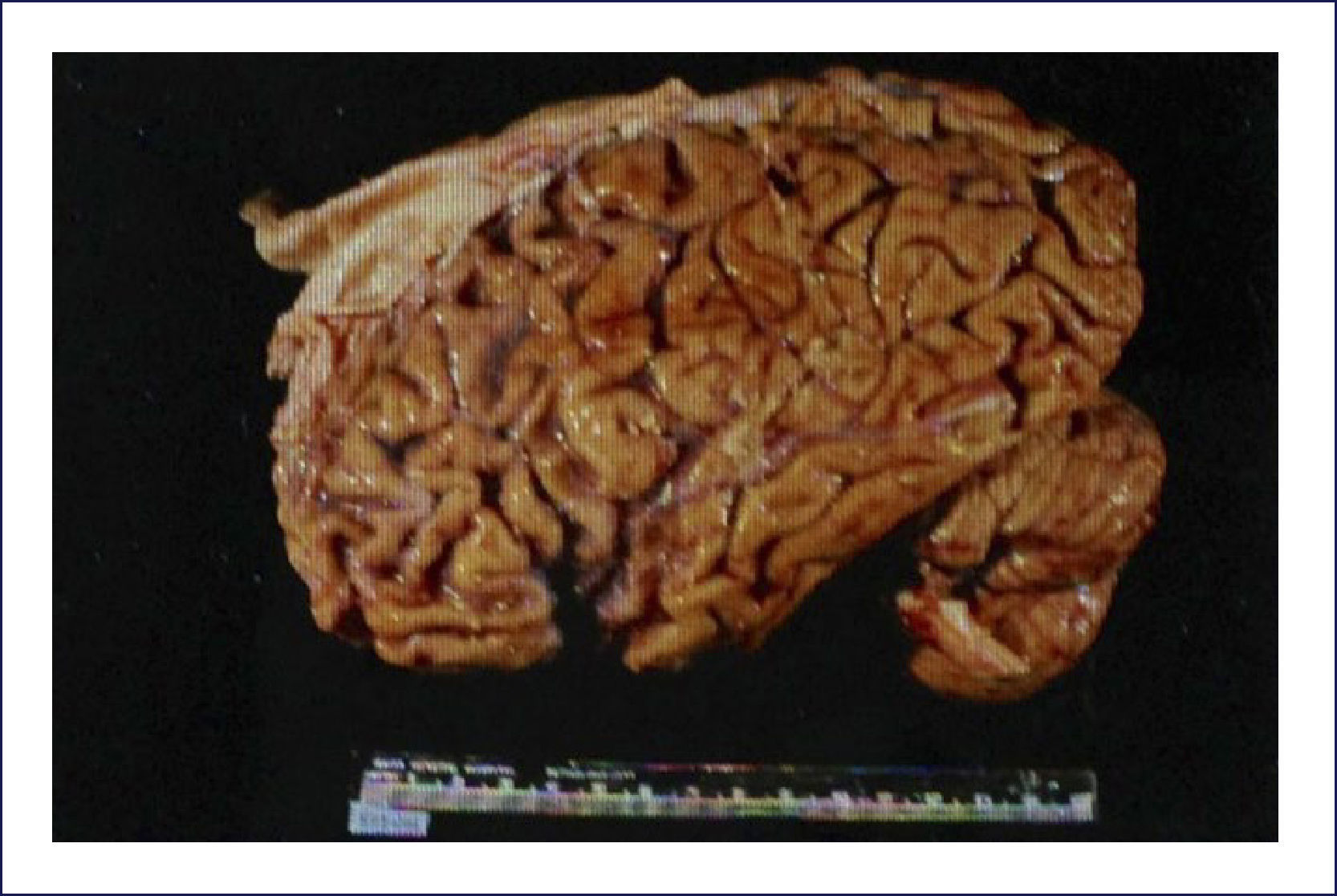
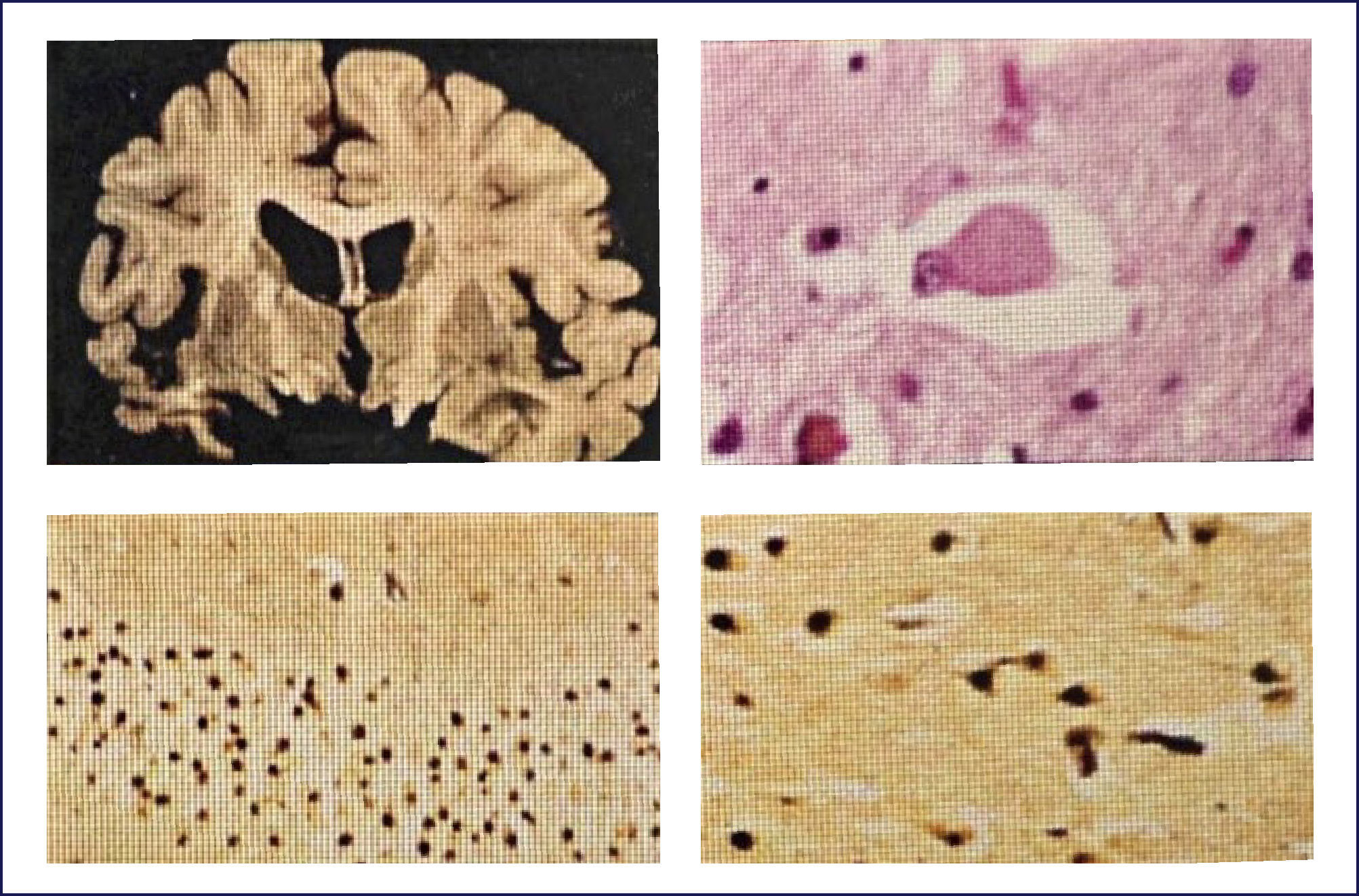
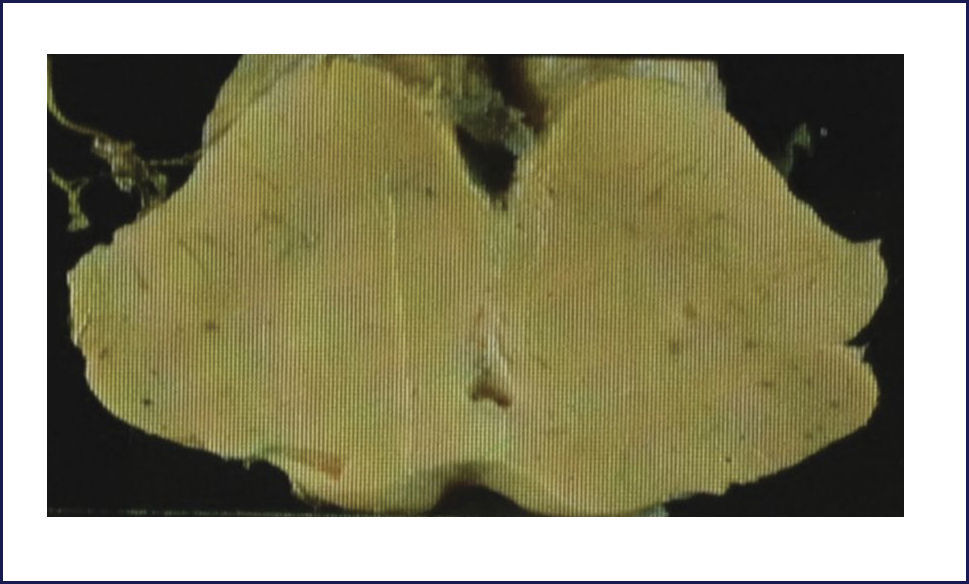
Enfermedad de Pick: Es una demencia esporádica, que característicamente comienza en la quinta o sexta década de la vida. Los cambios conductuales son severos. El patrón clínico de los síntomas corresponde a la distribución de las lesiones, las que comprometen los lóbulos frontales precozmente en el transcurso de la enfermedad. Macroscópicamente el encéfalo exhibe una severa atrofia circunscrita y mucho más evidente en los lóbulos frontales; aún cuando compromete los temporales, típicamente respeta el tercio posterior del giro temporal superior. Puede haber compromiso hipocampal y ello es responsable del trastorno de la memoria. La corteza parietal se afecta rara vez y la occipital siempre está respetada (Figura 7). Coincidente con este patrón de compromiso lobar, hay una mayor dilatación de la porción anterior de los cuernos frontales y temporales de los ventrículos laterales. Microscópicamente, las áreas corticales comprometidas muestran una severa pérdida neuronal, con una densa gliosis astrocitaria, generalmente acompañada con microvacuolización de la corteza. La enfermedad de Pick se caracteriza por la presencia de los cuerpos de Pick -inclusiones neuronales citoplasmáticas-, redondeadas, homogéneas, pálidamente visibles con la tinción de H&E. La inmunohistoquímica muestra que estas inclusiones contienen 3RTAU. Los cuerpos de Pick son fuertemente argirofílicos y se detectan muy bien usando las apropiadas tintines de plata (Figura 8). También son frecuentes de ver las neuronas balonadas, las que generalmente se conocen como células de Pick. Cuando están comprometidos los núcleos grises profundos, también hay importante pérdida neuronal y gliosis, pero no la acumulación de cuerpos de Pick. Estos cambios son más predominantes en la cabeza del caudado, aunque también pueden verse en el putamen, pallidum y tálamo 1,2.
.")
, y corteza cerebral (panel derecho) se tiñen marcadamente con las tinciones de plata. (Foto: colección personal).")
PANEL DE FOTOS ILUSTRANDO LA NEUROPATOLOGÿA DE LA ENFERMEDAD DE PICK
Superiores: Izquierda: corte transversal de cerebro que muestra la severa atrofia fronto temporal asimétrica y dilatación ventricular. Derecha: Cuerpo de Pick teñido con hematoxilina eosina, al interior de una neurona. Inferiores: Cuerpos de Pick en neuronas del giro dentado del hipocampo (panel izquierdo), y corteza cerebral (panel derecho) se tiñen marcadamente con las tinciones de plata. (Foto: colección personal).
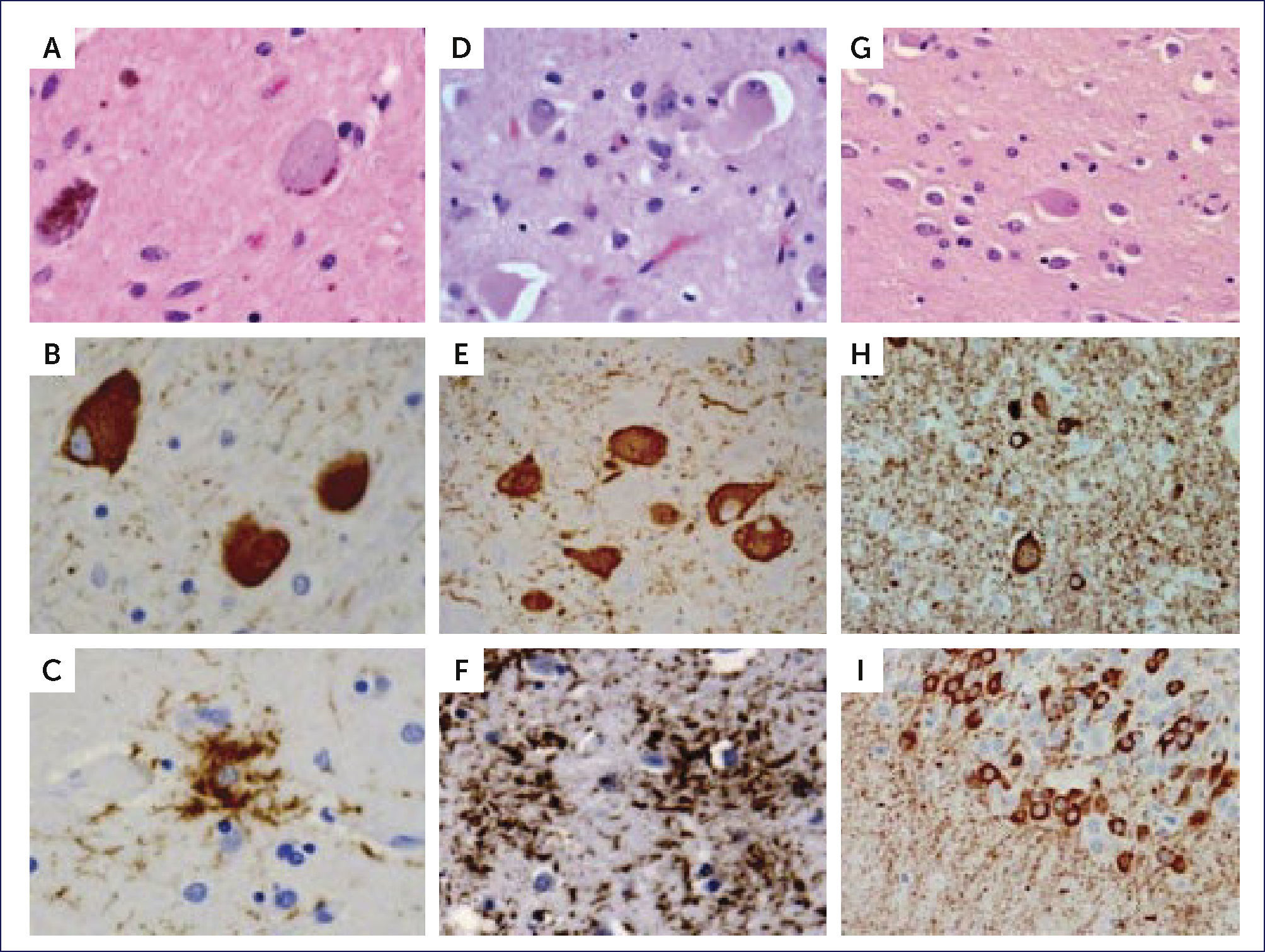
DLFT con Mutaciones MAP: En las formas familiares de DLFT-TAU se encuentra mutaciones del gen MAPT que codifica la proteína tau. Estas mutaciones se califican en aquellas que alteran el acoplamiento del mRNA para TAU y aquellas mutaciones puntuales. La alteración del acoplamiento produce una desviación del balance entre el TAU 3R y 4R. Se piensa que este desequilibrio contribuye a la iniciación de la disfunción celular. La enfermedad se caracteriza por atrofia difusa de los lóbulos frontal y temporales, correlacionada con las alteraciones cognitivas. Microscópicamente también hay extensa pérdida neuronal y astrocitosis en las áreas afectadas. Frecuentemente se encuentra inclusiones TAU en la glía. Las neuronas acumulan inclusiones TAU en una forma difusa, y se les llama pre-ovillos. En estas se encuentran isoformas de TAU3R, 4R o una combinación (Figura 9g y h) 1,2,10.
 y TAU (b) y astrocitos velludos con inmunohistoquímica para TAU (c). En la CBD: neuronas balonadas con H&E (d) pre-ovillos en el núcleo basal (e) y placas astrocíticas en la corteza (f), ambos con inmunohistoquímica para TAU. En la mutación MAPT: neuronas balonadas con H&E (g),neuronas balonadas, neuritas distróficas y pre-ovillos en la corteza (h) y pre-ovillos en el giro dentado (i) con inmunohistoquímica para TAU. (Foto: referencia bibliográfica 10).")
PANEL DE FOTOS CON LA HISTOLOGÿA DE PATOLOGÿAS MÿS FRECUENTES TAU 4R
En la PSP: ONF globosos con H&E (a) y TAU (b) y astrocitos velludos con inmunohistoquímica para TAU (c). En la CBD: neuronas balonadas con H&E (d) pre-ovillos en el núcleo basal (e) y placas astrocíticas en la corteza (f), ambos con inmunohistoquímica para TAU. En la mutación MAPT: neuronas balonadas con H&E (g),neuronas balonadas, neuritas distróficas y pre-ovillos en la corteza (h) y pre-ovillos en el giro dentado (i) con inmunohistoquímica para TAU. (Foto: referencia bibliográfica 10).
Algunos casos de DLFT no están asociados a patología de Pick ni mutaciones MAPT, aunque del punto de vista macro y microscópico no se pueden diferenciar de los casos con mutaciones.
DLFT con proteinopatía TDP-43 (DLFT-TDP)La DLFT con inclusiones TAU negativas, TDP-43 positivas da cuenta de aproximadamente la mitad de los casos de autopsia de DLFT confirmados. Varios locus genéticos albergan mutaciones causales para DLFT-TDP e incluye genes para TDP-43, progranulina y C9orf72. También pueden ocurrir formas esporádicas. En general, los tipos de inclusiones que definen los subtipos de DLFT no se correlacionan fuertemente con la presentación clínica. La DLFT-TDP a menudo se asocia a Esclerosis Lateral Amiotrófica, particularmente en los casos con mutaciones en TDP-43.
El examen macroscópico del encéfalo es similar a otras formas de DLFT. Los hallazgos microscópicos revelan espongiosis da las capas corticales superficiales, particularmente de la capa II de la corteza frontal. La TDP-43 es una proteína nuclear. La tinción de los núcleos neuronales es uniforme en la inmunohistoquímica. En la DFT-TDP la inmunohistoquímica muestra inclusiones intraneuronales positivas para ubiquitina y proteína TDP-43 de diverso tipo y distribución variable. Las inclusiones TDP-43 positivas neuronales se ubican en el cuerpo celular, en el núcleo o en las neuritas; también se observan inclusiones gliales. Las inclusiones predominan en las cortezas frontal y temporal, en el estriado y en el giro dentado del hipocampo.
Diferentes esquemas de clasificación se han propuesto para la DLFT-TDP, basados en el aspecto, abundancia y distribución de las inclusiones y también en un intento de tener alguna correlación con alteraciones genéticas, así menos con el fenotipo 2,10.
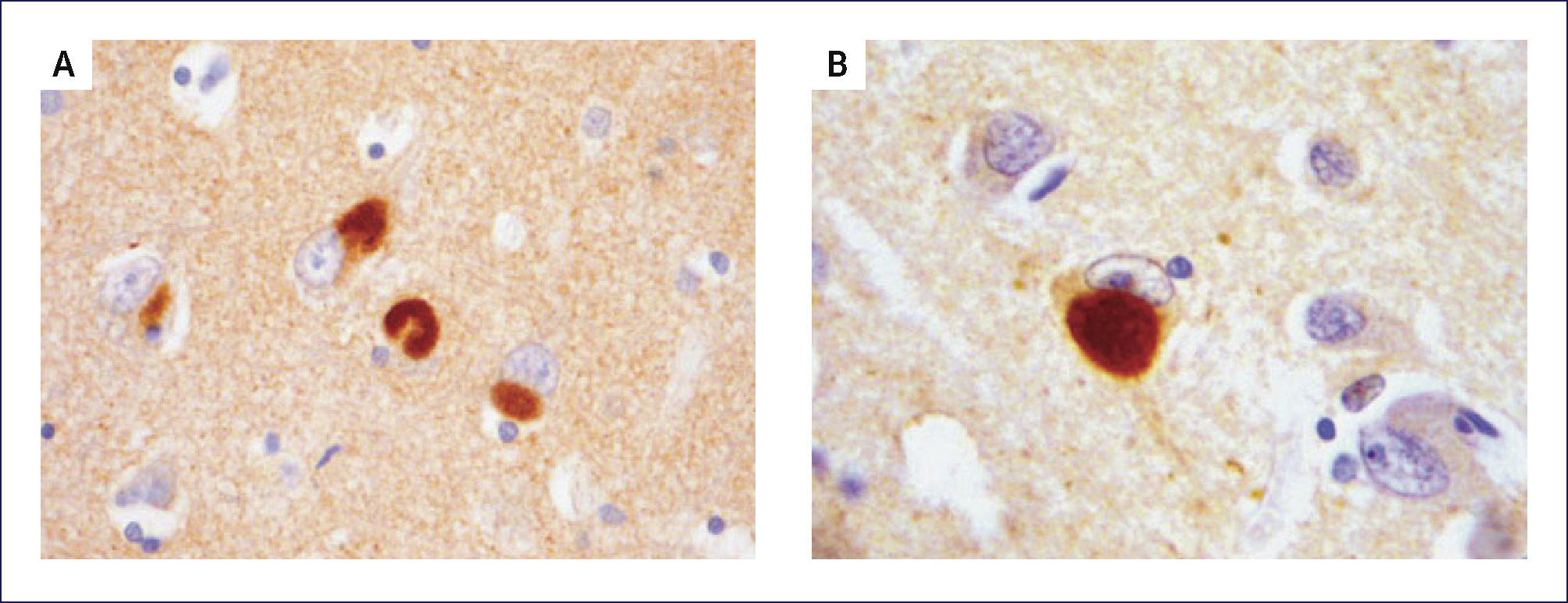
DEMENCIA CON CUERPOS DE LEWY (DCL)Los cambios neuropatológicos incluyen la presencia de cuerpos de Lewy y neuritas Lewy en la corteza cerebral así como en el tronco cerebral. A menudo se asocia a patología EA. Macroscópicamente la atrofia cerebral no es tan acentuada como en la EA con similar trastorno cognitivo. Se encuentra palidez de la sustancia nigra y del locus coeruleus como la que se ve en la EP. La atrofia del sistema límbico puede ser prominente. A nivel microscópico hay pérdida neuronal y gliosis generalmente restringida a regiones del troncoencéfalo, principalmente sustancia nigra y locus coeruleus. La severidad de la injuria es típicamente comparable a la observada en la EP, junto a la presenta de cuerpos de Lewy -inclusiones neuronales citoplasmáticas- eosinofílicas, laminadas. En contraste con estos hallazgos, la corteza cerebral, hipocampo y amígdala no se ven afectados con las tinciones de rutina. Sin embargo, con inmunohistoquímica con Ac en contra alfa-sinucleína, se revela extensamente la presencia de inclusiones en el citoplasma de las neuronas corticales, especialmente en las capas más profundas (particularmente temporal, insular y cingulada) (Figura 10). Estas también están acompañadas de neuritis de Lewy en la neuropila cortical y espongiosis de las capas más superficiales de la corteza cerebral. Se destaca también la presencia de los cuerpos de Lewy en la formación reticular del troncoencéfalo, junto a neuritis distróficas (neuritas de Lewy). En la amígdala se encuentra cuerpos de Lewy en las neuronas, mientras que en el hipocampo aparece solamente neuritis de Lewy en las regiones CA2-3. Como se mencionó previamente, muchos encéfalos con DCL también presentan placas y ONF, aunque generalmente no en la severidad que se encuentra en la EA 1,2.
PATOLOGÿA DE LAS DEMENCIAS DEGENERATIVAS CON TRASTORNO DEL MOVIMIENTOENFERMEDAD DE PARKINSON (EP) y para anti-ubiquitina (B).(Foto: tomada de referencia bibliográfica 1).")
El diagnóstico de EP se basa en la presencia de la tríada de parkinsonismo: temblor, rigidez y akinesia. La alteración neuropatológica más llamativa de la EP al estudio macroscópico del cerebro es la palidez de la sustancia nigra en el mesencéfalo, así como del locus coeruleus en la protuberancia (Figura 11). Microscópicamente se observa que las estructuras donde se observa palidez presentan pérdida neuronal y gliosis astrocitaria, especialmente en la parte compacta de la sustancia nigra. La neuromelanina se puede observar libre en la neuropila o al interior de los macrófagos. Las neuronas remanentes presentan cuerpos de Lewy en sus núcleos (Figura 12). Otras zonas habitualmente comprometidas son el núcleo dorsal del vago, el núcleo nasal de Meynert y la amígdala. Los cuerpos de Lewy también pueden estar presentes en la corteza y en los núcleos autonómicos de la médula espinal, ganglios, plexo pélvico, suprarrenales. La inclusiones de Lewy contienen alfa sinucleína.
.")
.")
PSP o Síndrome de Steele-Richardson-Olszewsky se caracteriza clínicamente por parkinsonismo asociado con parálisis supranuclear. El parkinsonismo generalmente no tiene temblor, se presenta con hipertonía, retrocolis (en vez de flexión como en la EP) y rigidez axial (más que compromiso de las extremidades como en la EP). La parálisis seudobulbar y los trastornos cognitivos que llevan a una demencia son frecuentes. Del punto de vista neuropatológico, macroscópicamente la anormalidad más característica es la atrofia del mesencéfalo y del tegmentum pontino. Comúnmente hay palidez de la sustancia nigra y del locus coeruleus, como la que se ve en la EP, pero con una atrofia variable del globus pallidus. Habitualmente la corteza cerebral no está afectada, aunque en casos con trastorno cognitivo severo puede haber atrofia frontotemporal. Del punto de vista microscópico, la característica diagnóstica de la PSP es la combinación de pérdida neuronal y gliosis astrocitaria, con acumulación neuronal y glial de proteína TAU, vista por la inmunohistoquímica. Hay extenso compromiso de la sustancia nigra, núcleos subtalámicos y globus pallidus, así como del troncoencéfalo: folículo superior, área pretectal, sustancia gris periacueductal, y la formación reticular mesencefálica y pontina. Además puede haber un compromiso moderado de los núcleos dentados el cerebelo, locus coeruleus, núcleos óculomotores, núcleos pontinos, formación reticular bulbar, complejo olivar inferior y tálamo. En las regiones afectadas, la pérdida neuronal y gliosis se asocia a inclusiones que contienen proteína TAU, tanto en las neuronas como en la glia. Al interior de las neuronas, la proteína tau forma ONF que tienen un característico aspecto globoso (Figura 13). Los astrocitos rizados, considerados altamente específicos de esta enfermedad, habitualmente son abundantes en el putamen, también se encuentran comúnmente en la corteza cerebral, especialmente área promotora del lóbulo frontal (Figura 9 a, b, c) 1,2,10.
.")
Casi todos los casos de PSP son esporádicos, aunque existen casos raros en los cuales ocurren mutaciones MAPT. Los agregados de TAU en neuronas y glía en la PSP están compuestos principalmente de las isoformas 4RTAU.
DEGENERACIÿN CORTICOBASAL (DCB)La DCB es una entidad clínico-patológica en la que hay degeneración de áreas corticales y de los ganglios basares (incluyendo la sustancia nigra). Está relacionada a la PSP, con la acumulación de 4RTAU en neuronas y glía, aunque se puede hacer distinción entre ambos desde el punto de vista clínico y neuropatológico, basado en la distribución distintiva y carácter de las lesiones.
La enfermedad se presenta con rigidez, inestabilidad, clonías de la extremidad superior y menos comúnmente la inferior. Hay asimetría en estos movimientos anormales, y es común el que los pacientes desarrollen una apraxia progresiva llamada ⿿extremidad ajena⿿, fenómeno en que la extremidad se mueve sin el control voluntario y en asociación con una sensación que no le pertenece al individuo. En algunos pacientes hay trastorno cognitivo con afasia y demencia del tipo frontotemporal. En algunos pacientes los trastornos cognitivos pueden ser más predominantes que el trastorno motor. Del punto de vista neuropatológico, macroscópicamente la atrofia cortical es el hallazgo típico, más prominente alrededor de la fisura de silvio o de distribución fronto temporal, a menudo asimétrica. La sustancia nigra está despigmentada y puede haber atrofia de los ganglios basales. Microscópicamente lo característico es la pérdida neuronal, gliosis astrocitaria e inclusiones de TAU 4R en neuronas y glía. Además, en la corteza cerebral es posible encontrar neuronas infladas que han perdido la sustancia de Nissl (acromáticas). En la sustancia nigra la pérdida neuronal se asocia a gliosis y las neuronas remanentes nigrales muestran prominentes ONF globosos. La inmunotinción de proteína TAU muestra reactividad en los ovillos de las neuronas así como en muchas neuronas infladas. La acumulación de proteína TAU en los astrocitos forma figuras distintivas en la sustancia gris, que se llaman placas astrocíticas: la proteína tau se acumula en el extremo distal de los procesos astrocíticos, mientras que el centro de la placa está desprovisto de inmunoreactividad TAU. Estas placas son muy conspicuas en la corteza y en el putamen (Figura 9 d, e, f) 1,2,10.
ATROFIA MULTISISTÿMICA (AMS)Como lo dice su nombre, en la AMS el proceso degenerativo cruza varios sistemas funcionales y por ende no encaja bien en una sola de las categorías clínicas presentadas. Frecuentemente tiene parkinsonismo y como en la EP, está incluida dentro de las sinucleopatías. Tres enfermedades, a saber la Degeneración Estriatonigral, la Ataxia Olivopontocerebelosa (OPCA) y el Shy Draeger, que originalmente se creía eran distintas, se reunieron bajo este nombre común, después de reconocer que los individuos afectados a menudo empezaban con un complejo sintomático, pero eventual y gradualmente adquirían los otros. Esta agregación clínica se validó posteriormente con el reconocimiento que los hallazgos neuropatológicos de cuerpos de inclusión distintivos en células gliales eran comunes a todos estos pacientes. Subsecuentemente se mostró que estas inclusiones contenían alfa-sinucleína, llevando a clasificar la AMS como una sinucleopatía, junto a la EP y a la DCL. No se ha encontrado mutaciones en el gen para la alfa sinucleína en la AMS, la que aparece existir sólo como una enfermedad esporádica 1,2.
Clínicamente la AMS puede mostrar predominancia de parkinsonismo (AMS-P) o de ataxia cerebelosa (AMS-C); es relativamente raro que la disfunción autonómica sea la única manifestación de la enfermedad, aunque algún tipo de disfunción autonómica es frecuente en la mayoría.
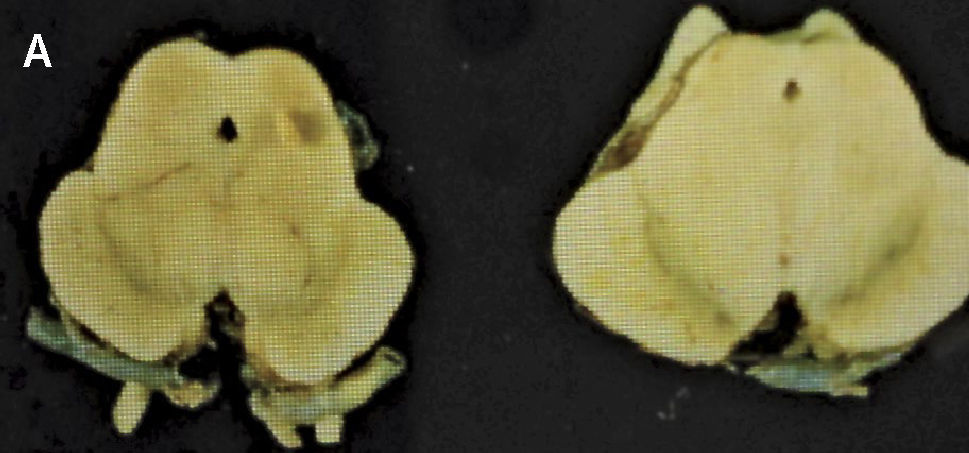
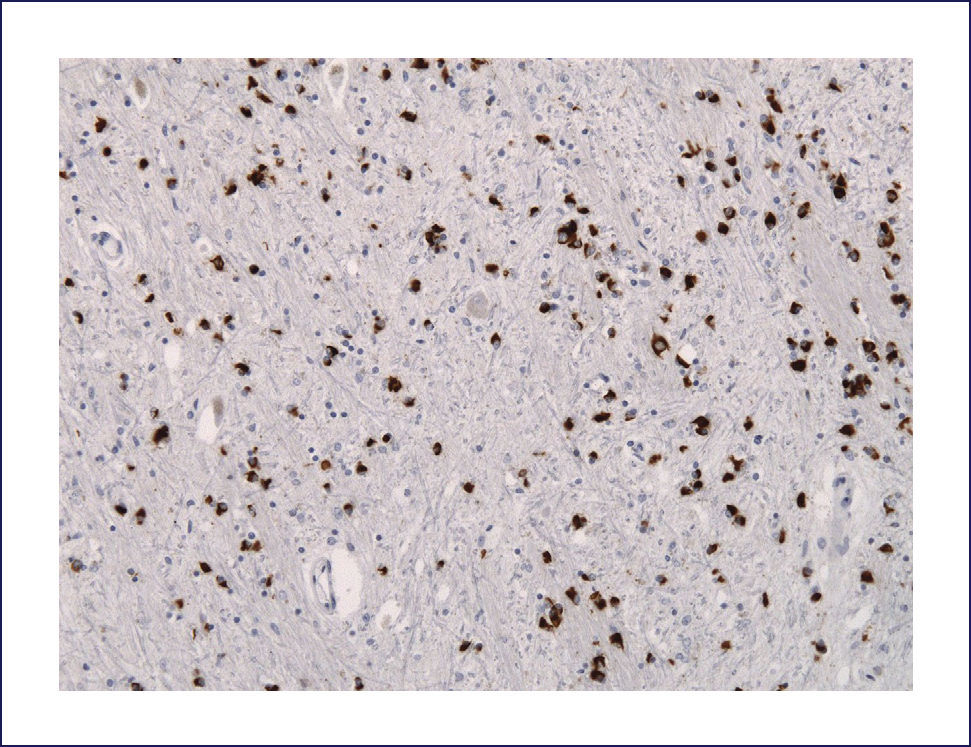
Como se puede esperar del espectro clínico, macroscópicamente puede haber un variado rango de anormalidades. El mejor correlato de la AMS-P es la combinación de palidez de la sustancia nigra con atrofia del putamen, a menudo asociado a una decoloración grisáceo-verdosa de éste último. Cuando el componente cerebeloso es prominente (AMS-C), el cerebelo, la protuberancia y el complejo olivar inferior están atróficos (Figuras 14 a, b, c). Microscópicamente hay pérdida neuronal y gliosis de las áreas comprometidas. Con las tinciones habituales de hematoxilina eosina no se evidencia mucho más. Las tinciones de plata o inmunohistoquímica para alfa sinucléina y ubiquitina mostrarán las inclusiones citoplasmáticas gliales características en los oligodendrocitos. Están ampliamente distribuidas en el encéfalo y aparecen como estructuras crescénticas, particularmente envolviendo el núcleo extendiéndose lejos de este 11 (Figura 15).
, correspondiente a AMS. (Foto: colección personal). B: Corte transversal de protuberancias fijadas en formalina, ilustrando marcada atrofia (izquierda), correspondiente a AMS. (Foto: colección personal). C: Corte transversal de bulbos fijados en formalina, ilustrando marcada atrofia (izquierdo) correspondiente a AMS. (Foto: colección personal).")
, correspondiente a AMS. (Foto: colección personal). B: Corte transversal de protuberancias fijadas en formalina, ilustrando marcada atrofia (izquierda), correspondiente a AMS. (Foto: colección personal). C: Corte transversal de bulbos fijados en formalina, ilustrando marcada atrofia (izquierdo) correspondiente a AMS. (Foto: colección personal).")
, correspondiente a AMS. (Foto: colección personal). B: Corte transversal de protuberancias fijadas en formalina, ilustrando marcada atrofia (izquierda), correspondiente a AMS. (Foto: colección personal). C: Corte transversal de bulbos fijados en formalina, ilustrando marcada atrofia (izquierdo) correspondiente a AMS. (Foto: colección personal).")
A: Corte transversal de mesencéfalos fijados en formalina, ilustrando marcada atrofia (izquierdo), correspondiente a AMS. (Foto: colección personal). B: Corte transversal de protuberancias fijadas en formalina, ilustrando marcada atrofia (izquierda), correspondiente a AMS. (Foto: colección personal). C: Corte transversal de bulbos fijados en formalina, ilustrando marcada atrofia (izquierdo) correspondiente a AMS. (Foto: colección personal).
.")
Esta breve revisión sobre los aspectos neuropatológicos de las demencias neurodegenerativas ilustra alguno de los aspectos comunes de la fisiopatología de estas enfermedades, tan variadas del punto de vista clínico y patológico y como la contribución de la patología molecular y la genética han permitido avanzar en esta nueva forma de relacionarlas. En la actualidad, la combinación de la neuropatología clásica, con la inmunohistoquímica, biología molecular y genética es la metodología necesaria para caracterizar estas patologías.
La autora declara no tener conflictos de interés, en relación a este artículo.