



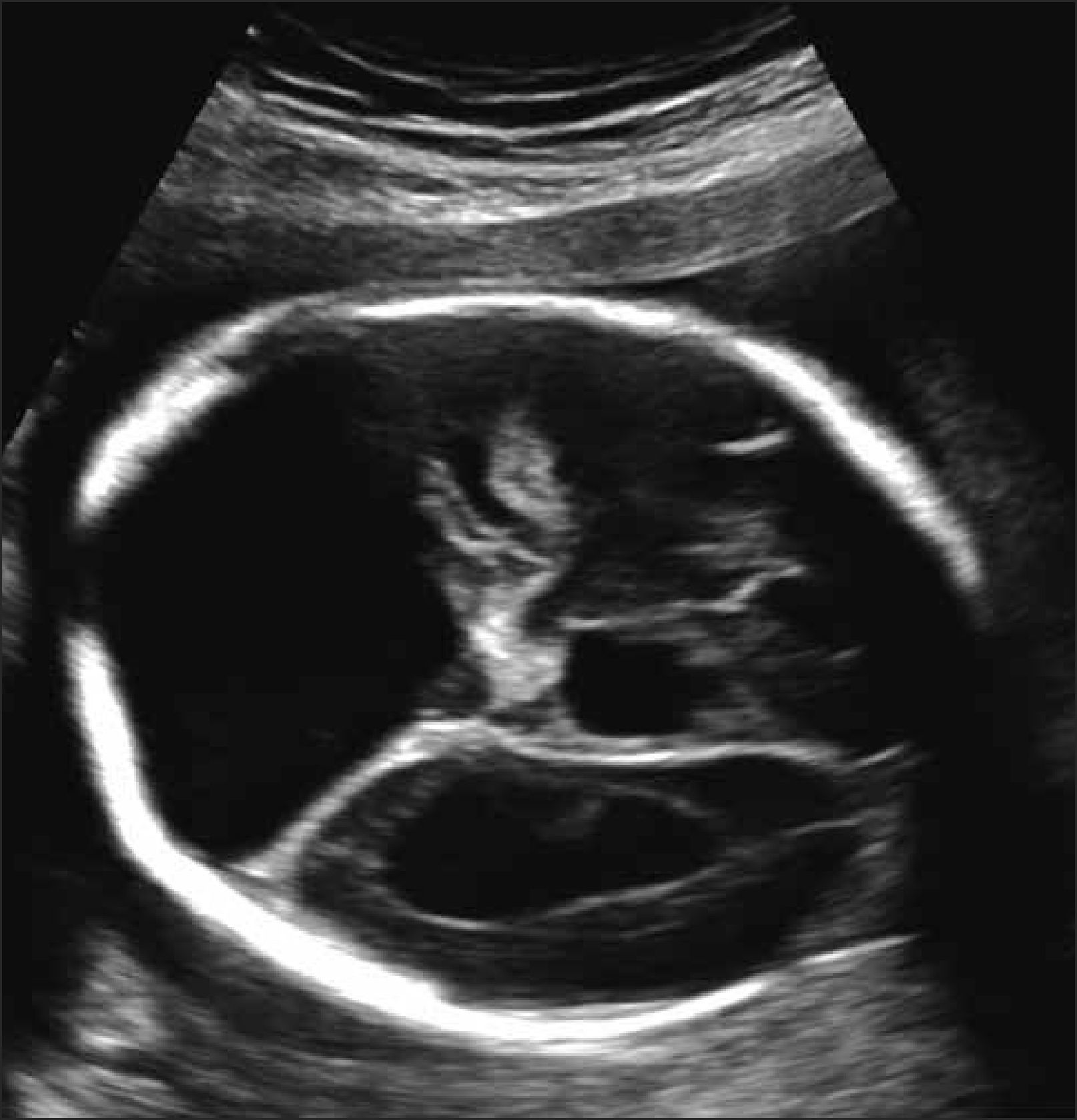

El diagnóstico y manejo prenatal de enfermedades raras involucra un estudio multidisciplinario. Desde una visión obstétrica, herramientas de imagenología como la ultrasonografía y en menor medida la resonancia magnética fetal (RMF) son esenciales para el diagnóstico de anomalías morfológicas y sospecha de defectos cromosómicos. Para el diagnóstico de enfermedades cromosómicas el estudio de cariograma obtenido de vellosidades coriales, líquido amniótico o sangre fetal mediante técnicas invasivas tal como biopsia corial, amniocentesis o cordocentesis ha sido hasta hace poco el gold standard del diagnóstico. Nuevas técnicas moleculares capaces de detectar microdeleciones como es el microarray nos ha permitido aproximarnos al origen de las enfermedades raras. Se revisarán además algunos de los defectos anatómicos raros y su enfoque neonatal.
Prenatal diagnosis and management of rare diseases is a multidisciplinary task. From the obstetric vision, prenatal diagnosis is based on fetal images obtained by ultrasound or magnetic resonance and fetal chromosomes study. The study of fetal morphology allows us the diagnosis of fetal abnormalities and the suspicion of chromosomal defects. Fetal chromosomal study is obtained either by chorionic villus sampling, amniocentesis and cordocentesis, all associated to fetal risk. New screening techniques such as fetal DNA on maternal blood or the study of fetal micro deletions using micro Array on amniotic fluid has extended diagnostic opportunity of rare fetal diseases. We will review some of the most common rare diseases and the neonatal approach.
Durante el período perinatal, el paciente portador de una enfermedad rara requiere, tanto para su diagnóstico como para su manejo de un enfoque multidisciplinario que además del obstetra, involucra a genetistas, radiólogos, cirujanos infantiles, pediatras neonatólogos y psicólogos, entre otros. Sin este enfoque, el diagnóstico, manejo y consejo se hace extremadamente difícil y más aún, deficiente. La evaluación prenatal es la primera aproximación a un defecto fetal. Es en esta etapa en la que se aproxima el diagnóstico y etiología más probable de la enfermedad, junto con trazar un plan de seguimiento y modo de nacimiento. Si bien la mayoría de los defectos no tiene un tratamiento prenatal, el preparar al equipo médico para recibir a un neonato con problemas es de los elementos que mejoran la condición postnatal y facilitan los cuidados futuros. Desde el punto de vista parental, el condicionar a los padres entregando las herramientas para entender la enfermedad y aconsejar desde el punto de vista humano y médico facilita la tarea del equipo.
Parte de la tarea diagnóstica involucra estudio imagenológico como ultrasonografía y resonancia nuclear magnética. Es en esta área donde el trabajo en equipo entre obstetra y radiólogo cobra importancia para llegar a un diagnóstico común.
El estudio cromosómico involucra muestra de tejido, ya sea biopsia corial, líquido amniótico o sangre fetal. Hasta hace poco, el gold standard era el cariograma capaz de evidenciar defectos estructurales mayores de los cromosomas como trisomías, deleciones, duplicaciones y traslocaciones. En los últimos años, avances tecnológicos han permitido mediante técnica de micro-array detectar microdeleciones o pequeños variantes del cariograma, responsables de síndromes genéticos raros o por reconocer en el futuro. Esto último ha ampliado nuestras capacidades diagnósticas, pero también las incógnitas sobre la traducción clínica de las múltiples variantes del cariograma humano. Aquí cumple un rol esencial el médico genetista, capaz de orientar al equipo médico acerca de la indicación de estudio pre o postnatal y las posibles consecuencias del resultado de estudio genético.
Con toda esta información en conjunto con neonatología se decide la vía de parto, edad gestacional de nacimiento y complejidad de cama de recién nacido. Todo para lograr la menor morbimortalidad perinatal.
I.- Diagnóstico prenatal con ultrasonografíaEl diagnóstico prenatal se basa en la ultrasonografía. Es la primera aproximación al diagnóstico y sospecha de defectos fetales.
a) Evaluación a las 11 - 14 semanas. Esta ecografía, también llamada “Screening de Primer Trimestre” se realiza entre las 11/0 semanas y 14/0 semanas, fecha en que el feto mide entre 45 y 85mm de longitud céfalo nalgas (LCN). En ésta, se evalúa la anatomía fetal y marcadores de defectos cromosómicos como son la traslucencia nucal (TN), hueso nasal (HN), presencia de reflujo en la válvula tricúspide (RT) y flujo del ductus venoso (DV).
Excluyendo el examen del corazón fetal, la probabilidad de realizar una adecuada evaluación anatómica a esta edad gestacional es de un 96% 1.
En la mayoría de los estudios realizados entre las 11-14 semanas en población de alto y bajo riesgo con el objetivo de determinar la factibilidad de detectar malformaciones en primer trimestre, la prevalencia de una malformación mayor fue de un 0.6% (1.0-2.8). La detección de defectos mayores fue en promedio de un 52%, con un intervalo desde 18% hasta un 83% de detección 2–5.
La traslucencia nucal se refiere a la imagen traslucida en la región posterior al cuello fetal que se encuentra entre la piel y la base del cráneo. Si la medida de ésta se encuentra sobre el percentil 95 para la edad gestacional (mayor a 3,5mm), los riesgos de defectos anatómicos y cromosómicos son mayores.
La sensibilidad para la detección de defectos cromosómicos varía entre un 70-90%, dependiendo de la experiencia del operador, marcadores de defectos cromosómicos evaluados y uso de bioquímica materna (PAPP-A, fracción libre de BhCG). La mejor sensibilidad se da para el tamizaje de Síndrome de Down, que se presenta en 1 de cada 450 embarazos y es algo menos para la trisomía 18 (Síndrome de Edwars, 1 en 4.500), trisomía 13 (Síndrome de Patau, 1 en 5.000) y monosomía X (Síndrome de Turner, 1 en 3.000).
En base a un modelo de riesgos relativos que combina la edad materna y la presencia o ausencia de marcadores de defectos cromosómicos, se puede expresar este riesgo en un valor numérico que va de 1 en 2 (50% de probabilidad) a 1 en 20.0000. Es con esta información que se discute con los padres la opción de confirmación de defectos cromosómicos.
b) Ecográfica a las 20 – 24 semanas. También llamada ecografía morfológica. Este estudio es el de elección para el diagnóstico de las malformaciones fetales y anexos ovulares (cordón y placenta). Aun cuando la sensibilidad para la detección de defectos anatómicos en diversos estudios no supera el 50%, la detección de anomalías en unidades especializadas y personal calificado varía desde un 80 a 90% 6.
II. Diagnóstico prenatal con resonancia magnética fetalLa Resonancia Magnética Fetal (RMF) es una técnica no invasiva, que no utiliza radiación ionizante. La resonancia obtiene su imagen de la estimulación y posterior relación de protones de átomos de hidrógeno en un campo magnético de alta intensidad.
a) Aspectos generales y seguridad de la técnica
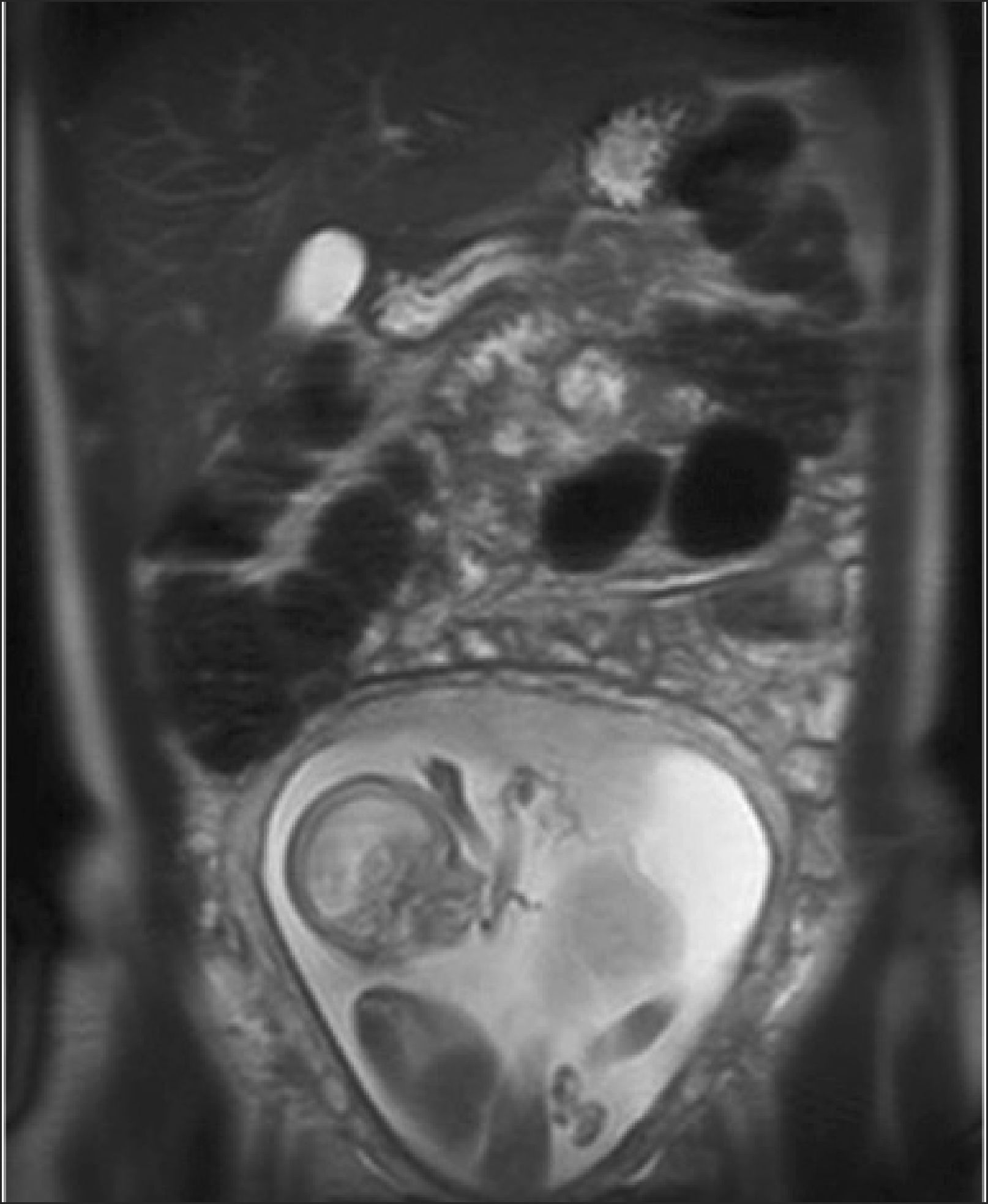
Durante la vida intrauterina, es posible obtener imágenes con fines diagnósticos dada la disponibilidad de adquisiciones rápidas, que permiten la representación del feto en diferentes planos, a pesar de los movimientos tanto fetales como del líquido amniótico (Figura 1).

Los cambios que se producen en la relajación de los protones de acuerdo a los diferentes tejidos desde donde se obtiene la señal que forma la imagen, son responsables de la gran resolución de contraste de las imágenes. Así, precisamente por el alto contenido de agua del feto es posible obtener gran detalle tanto del encéfalo y médula como del tronco y extremidades. Hasta ahora no han sido reportados efectos adversos de la técnica y su uso se ha extendido con las respectivas autorizaciones en EE.UU. (FDA) y de la Comunidad Europea. Los potenciales efectos relacionados a aumento de temperatura y la exposición a altos decibeles de ruido producidos por algunas secuencias, son atenuados por el abdomen materno 7. A pesar de estas consideraciones, en nuestro centro el examen se realiza durante el segundo trimestre, principalmente entre 28 – 32 semanas, lejos de la organogénesis y con mejor calidad en las imágenes debido al mayor tamaño del feto.
b) Indicaciones
A pesar su creciente uso, la resonancia magnética es un examen de segunda línea en la evaluación por imágenes del feto. Inicialmente se fue posicionando como técnica alternativa en fetos de difícil visualización en ecografía, como la escasez de líquido amniótico, la obesidad materna o la sospecha de malformaciones encefálicas en fetos con calotas ampliamente osificadas o definitivamente encajados en la pelvis materna. Más tarde, se fue sumando como complemento a la ecografía para confirmar o descartar diagnósticos que determinaran cambios de conducta. Recientemente, y en forma muy especialmente relacionada a la sospecha de enfermedades raras, la resonancia ha ido adquiriendo un importante rol para aportar o refinar hallazgos que permitan establecer asociaciones sindromáticas que permitan preparar al equipo obstétrico y neonatal para la llegada de un paciente con sospecha de una patología poco frecuente, tanto en el enfrentamiento inmediato del recién nacido frente a condiciones que amenazan la vida y a la contención de la familia cuando el diagnóstico perinatal es desfavorable 8.
c) Alteraciones generales y específicas
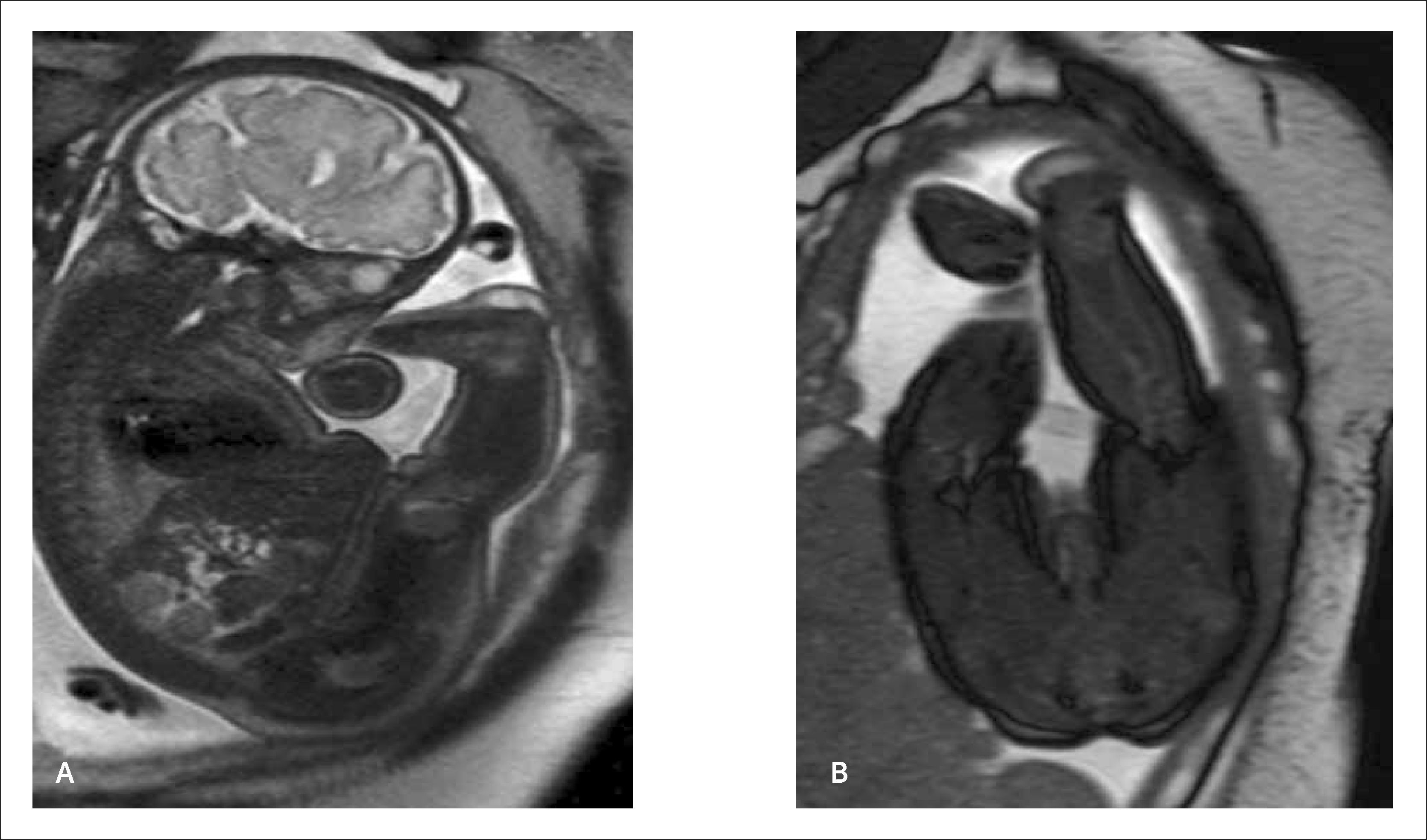
Los pacientes portadores de enfermedades raras pueden llegar a estudio, en general, en dos escenarios. Existe un grupo donde la afección de base es sospechada por ultrasonido y la RMF viene a confirmar los hallazgos y a buscar, específicamente, las alteraciones estructurales que completan el síndrome, como es el caso de la Malformación de Chiari tipo II que afecta la fosa posterior en paciente con Disrafia Espinal (Figura 2 a y b) o la desproporción de segmentos en pacientes con extremidades cortas y sospecha de displasia esquelética (Figura 3 a y b). En otros casos, si bien existe la sospecha fundada de una alteración genética o sindromática, el conjunto de múltiples hallazgos no logran encaminar el diagnóstico. En estos casos el detallado análisis anatómico de cada uno de los sistemas y las técnicas avanzadas como la volumetría, la espectroscopía o la tractografía, aportan información que permitirá redondear la sospecha y plantear un diagnóstico.
. Imagen T2 sagital (A) y T2 axial de extremidades inferiores (B).")
 y parasagital izquierda (B).")
La posibilidad cierta de contribuir en el diagnóstico y de influir en el manejo perinatal tiene directa relación con el traspaso de la información existente, tanto clínica, como de ultrasonidos previos realizados al paciente. Esto subraya el enfoque multidisciplinario y traslada las discusiones que cambiarán conductas a etapas cada vez más tempranas de la vida intrauterina.
IV. Diagnóstico prenatal con estudio cromosómicoLa indicación de estudio cromosómico involucra diversos aspectos que se relacionan con hallazgos prenatales y la decisión de los padres.
La técnica utilizada entre las 11 y 16 semanas para obtener una muestra para estudio cromosómico es la biopsia corial, técnica invasiva que mediante visión ecográfica y una aguja de 18 f se obtiene 1 a 5mg de vellosidad corial. Esta se hace vía abdominal o trasvaginal y se describe su asociación con pérdida fetal en un 1%.
Después de las 16 semanas y hasta término, el estudio es una amniocentesis. En esta se obtiene entre 5 a 20 cc de líquido amniótico. Nosotros recomendamos este procedimiento entre las 16 y 24 semanas y después de las 32 semanas. Esto porque la asociación a 1% de rotura prematura ovular antes de las 32 semanas empeora el pronóstico del embarazo independiente si hay o no un defecto asociado. El cariograma de líquido amniótico requiere el cultivo en laboratorio de amniocitos (células del amnios) y en promedio el cariograma tarda cuatro semanas. Técnicas más rápidas como FISH de cromosomas 21, 13 y 18 tardan 24 – 48 horas, pero elevan el costo del examen y no entregan información de todos los cromosomas.
La cordocentesis también se asocia a 1% de riesgo y se realiza desde las 20 semanas en adelante. Mediante visión ecográfica se obtiene una muestra sanguínea del cordón umbilical. El único beneficio es que el cariograma puede estar listo a la semana de tomada la muestra.
Hay dos modalidades de tamizaje de enfermedades cromosómicas.
1) Estudio 11- 14 semanas y biopsia corial para los grupos de riesgo elevado (<1 en 100). Este modelo tiene una sensibilidad de 95% aproximadamente para una tasa de 5% de falsos positivos. Esto significa que para detectar el 95% de los fetos portadores de Síndrome de Down, es necesario ofrecer un estudio cromosómico a 5% de la población, con un riesgo de 1% de muerte fetal asociado al estudio 9.
2) DNA libre en sangre materna (DNA sm). Esta técnica no invasiva es capaz de detectar en una muestra de sangre materna a partir de la novena semana de embarazo la fracción libre de DNA fetal, la que se obtiene mediante secuenciación masiva. El examen se expresa como, alto riesgo o bajo riesgo de defecto cromosómico.
Se han estudiado tres modelos de screening con esta técnica:
1.-Ecografía 11-14 y si el riesgo es de 1 en 100 o mayor, se ofrece DNA sm. Si el riesgo es alto se realiza una biopsia corial. Con este modelo se pesquisa el 86% de los fetos con Sd de Down, 88% de los fetos con Trisomia 18 y 13, con una tasa de falsos positivos del 0.4%
2.-Tamizaje según grupos de riesgo. Si el riesgo ecográfico es de 1 en 10 o mayor, se realiza biopsia corial, si el riesgo es entre 1 en 10 a 1 en 2500, se realiza DNA sm y si este es de alto riesgo se realiza biopsia corial. Si el riesgo ecográfico es menor a 1 en 2500 no se realiza más exámenes. Con esta estrategia se detecta al 98% de las trisomías 21, 18 y 13 con una tasa de falsos positivos de 0.8%
3.-Tamizaje a toda la población con DNF sm y biopsia corial a los catalogados con riesgo alto. La sensibilidad para t21 y t18ha sido del 100% con una tasa de falsos positivos del 0,3% y 0,2%. 10–11. Por ahora el inconveniente de generalizar este tamizaje es su alto costo, que bordea los 800 – 1000 dólares.
Solicitud de examen cromosómico en base a hallazgos en ecografía de 2do y 3er trimestre.
Las indicaciones para un estudio cromosómico en 2do y 3er trimestre son por solicitud paterna o por sospecha de defectos cromosómicos.
Se sugiere estudio cromosómico frente a las siguientes alteraciones, especialmente si se trata de defectos múltiples. (Ver Tabla 1).
Alteraciones asociadas con anomalías cromosómicas
| Malformaciones del SNC | Ventriculomegalia; Sd. Dandy Walker; Agenesia del cuerpo calloso; holoprosencefalia |
| Tórax | Hernia diafragmática |
| Corazón | Defectos cono-truncales; Canal AV |
| Abdomen | Onfalocele |
| Renal | Megavejiga |
| Esquelético | Sospecha de displasia esquelética |
Solicitud examen cromosómico en base a antecedentes familiares o hijos previos
Esta decisión se toma en base al consejo genético discutido previamente con genetista, y está basado en el riesgo de recurrencia, severidad de la enfermedad y consideración de los padres. Nosotros no recomendamos el estudio cromosómico si existe antecedente de un hijo con trisomía 13, 18, 21 y la evaluación del embarazo presente es normal.
Para otras enfermedades o síndromes malformativos infrecuentes, es importante conocer la mutación del caso índice y si se dispone de estudio de ese gen o conjunto de genes causantes de una enfermedad. En este caso y aconsejando a los padres en relación a riesgos asociados al estudio invasivo se toma la decisión caso a caso.
IV.- Malformaciones raras con diagnóstico mediante ultrasonografía y RMFEntre las malformaciones raras, es decir con frecuencias menores a 1 en 10.000 recién nacidos vivos diagnosticables en esta ecografía están:
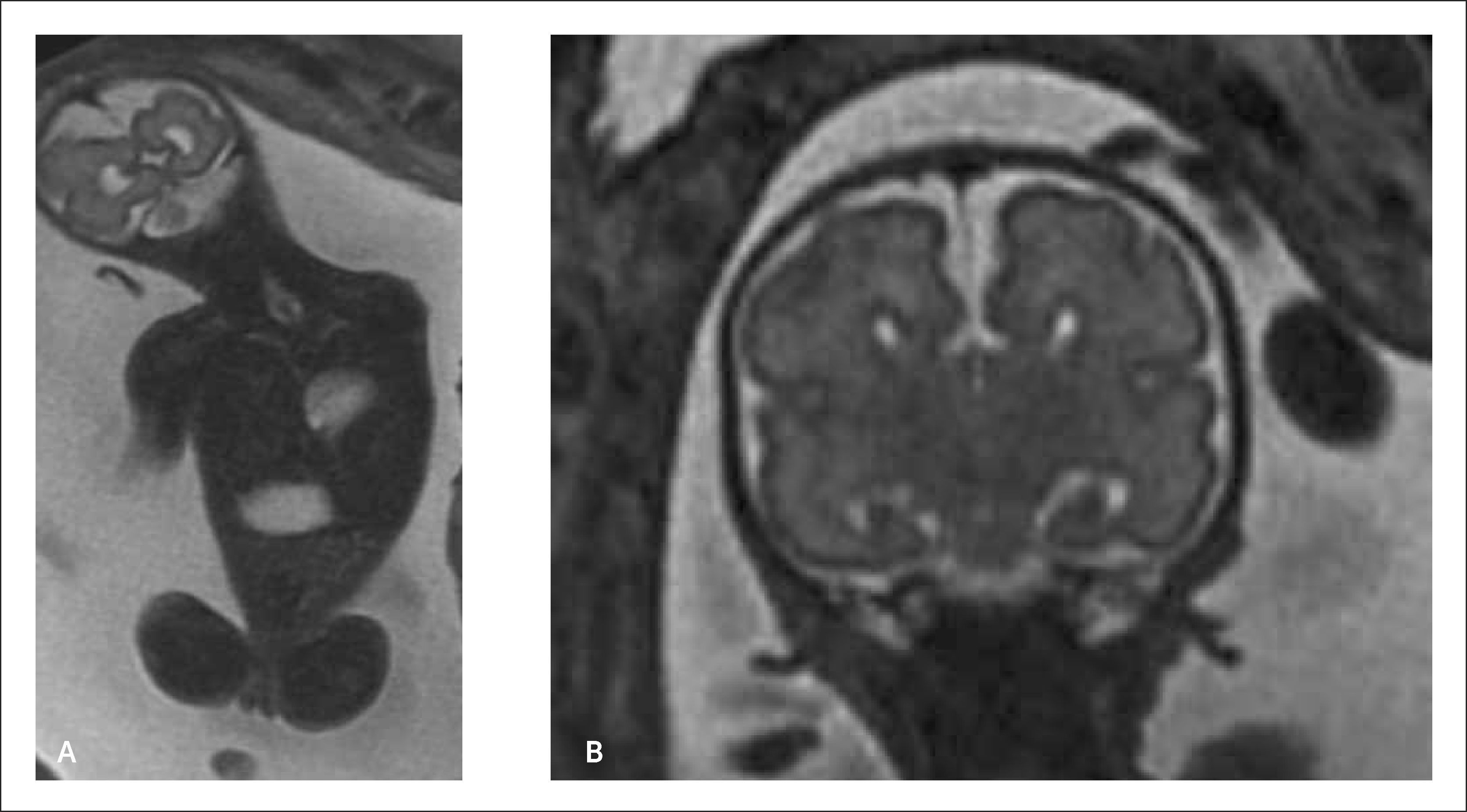
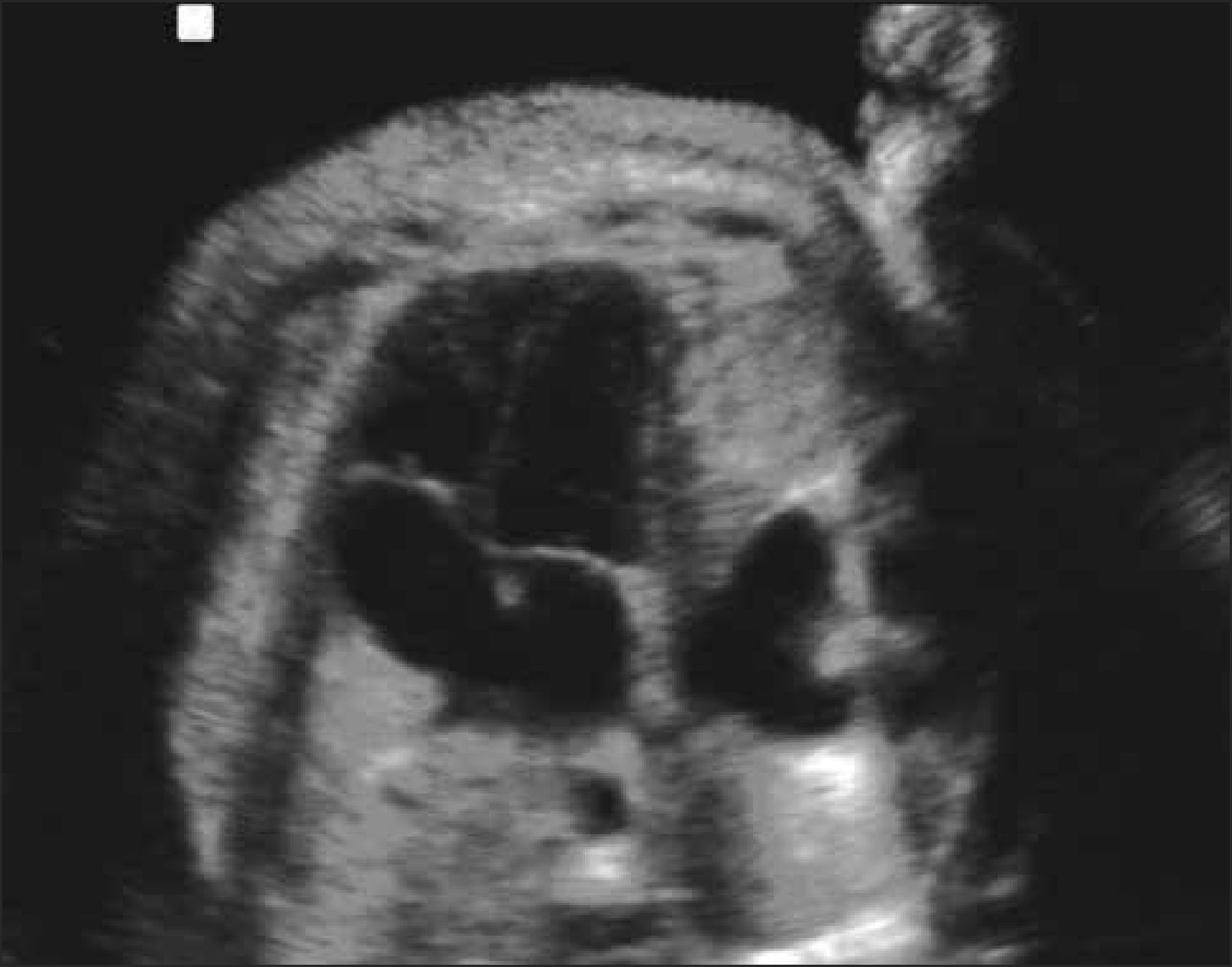
Holoprosencefalia: Se presenta en 1 de cada 10.000 embarazos. Se produce por una alteración en la segmentación del prosencéfalo en la etapa embrionaria. Su etiología puede ser de origen cromosómico, (trisomía 13 o 18), genético o en la mayoría de los casos de etiología desconocida. Se clasifica en alobar, lobar y semilobar dependiendo del grado de severidad. En su grado más severo (alobar), el diagnóstico no es difícil y marca un importante pronóstico fetal. La ausencia de la imagen clásica descrita como “alas de mariposa” que corresponde a los plexos coroideos en el plano transventicular del cerebro hace el diagnóstico a las 11- 14 semanas y la visualización de un quiste único central sin segmentación central y grados variables de fusión o segmentación del tálamo con fosa posterior normal hace el diagnóstico en la ecografía de 20 a 24 semanas.
La holoprosencefalia con defectos faciales se asocia en un 50% con aneuploidías, particularmente con trisomía 13. Se describen defectos genéticos de transmisión autonómica dominante y recesiva así como mutaciones puntuales de algunos genes (SHH, TGIF, ZIC2 y SIX3). La holoprosencefalia suele formar parte de los siguientes síndromes genéticos: DiGeorge, Meckel, Kallman, Smith-Lemli-Opitz, displasia campomelica, Hall-Pallister y Vasadi. Se han descrito tanto variantes con herencia autonómica dominante, autonómica recesiva y raramente ligada al cromosoma X. El riesgo de holoprosencefalia está aumentado 200 veces en hijos de madres con diabetes insulinodependiente. En la mayoría de los casos la etiología es desconocida. Para casos esporádicos, no cromosómicos, la recurrencia clásicamente reportada es de un 6%. El pronóstico de este defecto es variable siendo letal para las alobares y semilobar. La holoprosencefalia lobar se asocia a retraso mental de variable severidad, epilepsia, espasticidad, distonía y trastornos endocrinos tales como la diabetes insípida y deficiencia de hormona de crecimiento 12–19. Figura 4.
 Se observa una imagen anormal de los plexos coroideos.")
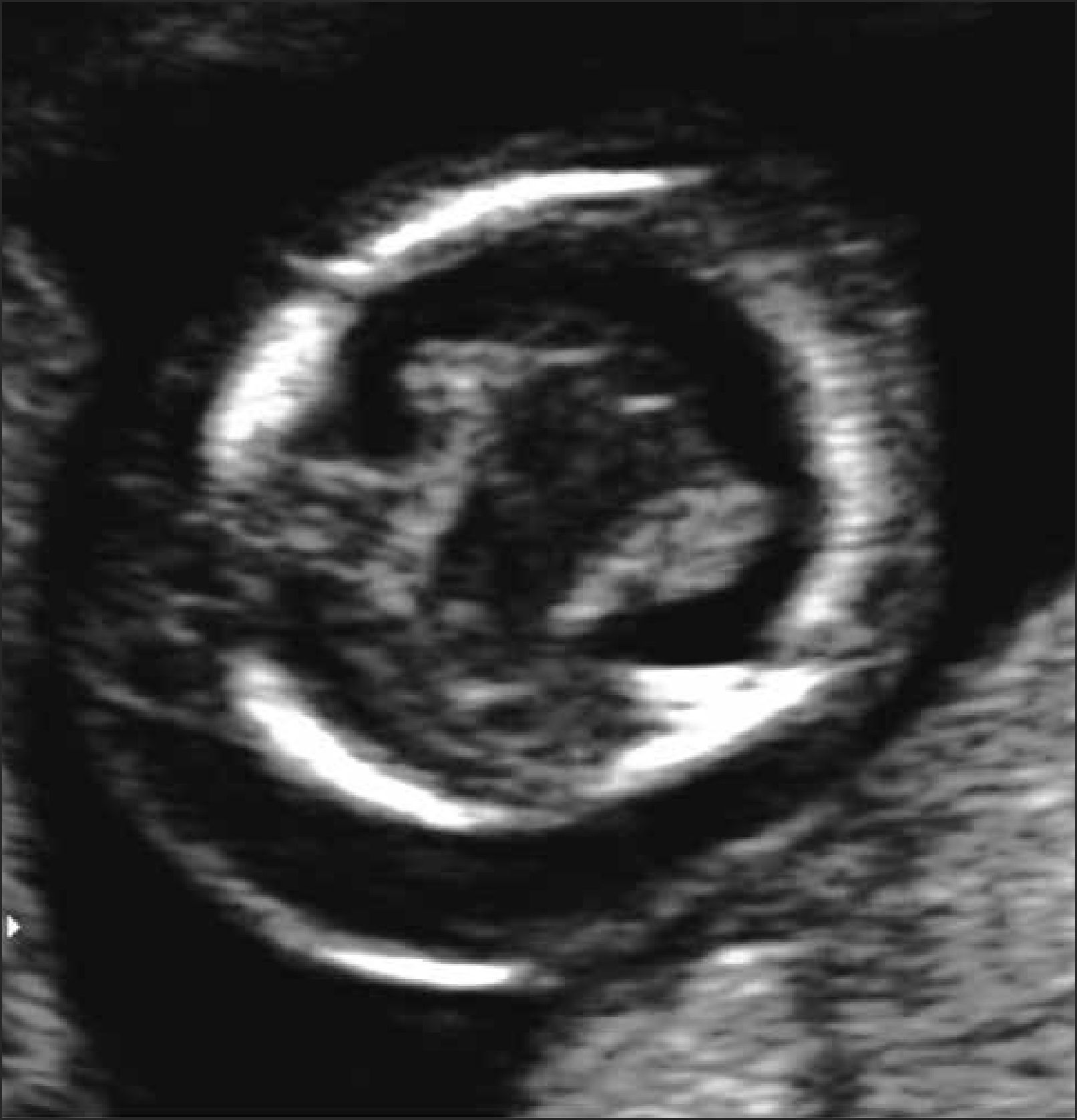
Complejo de Dandy-Walker. Se presenta en 1 de cada 30.000 nacidos vivos. El término Síndrome de Dandy-Walker(CDW) fue originalmente introducido para indicar la asociación de ventriculomegalia de grado variable, cisterna magna aumentada de tamaño y defecto en el vermis cerebeloso a través del cual un quiste se comunica con el 4to ventrículo. Con la introducción de la RMF, apareció el término Complejo Dandy-Walker, que distingue entre:
- a.
Dandy-Walker clásico. Fosa posterior aumentada de tamaño, agenesia total o parcial del vermis cerebeloso y elevación del tentorio (Figura 5).
- b.
Variante de Dandy-Walker. Hipoplasia variable del vermis cerebeloso con o sin aumento de tamaño de la cisterna magna.
- c.
Megacisterna Magna (habitualmente>10mm). Aumento de tamaño de la CM con integridad tanto del vermis cerebeloso como del 4to ventrículo.

Sin embargo y dada la dificultad de encasillar las alteraciones, éstas deben ser consideradas como parte de un continuo, ya que en todos los casos es posible encontrar algún grado de disgenesia del vermis, aún en los casos de megacisterna magna, mientras que el Dandy-Walker clásico y su variante tienen tantas similitudes que es imposible a menudo establecer una distinción precisa. Se asocia a trisomía 13,18 y triploidia, más de 50 síndromes genéticos, infecciones congénitas, teratógenos como warfarina o puede permanecer inexplicado. El CDW tiene un mal pronóstico aun cuando sea un defecto aislado. Tiene una alta mortalidad neonatal (alrededor de 20%) y la severidad del compromiso neurológico depende primordialmente de la integridad del vermis. La hipoplasia de este se asocia a secuelas neurológicas severas en el 85% de los casos 20–22.
Lesiones cerebrales destructivas. Se presentan en uno de cada 10.000 nacimientos. Dentro de ellas están:
- •
Hidranencefalia. Se produce por una lesión isquémica por obstrucción en el territorio de las carótidas internas, hidrocefalia prolongada o infecciones como toxoplasmosis o citomegalovirus.
- •
Porencefalia. Por infarto de las arterias cerebrales o hemorragias del parénquima cerebral.
- •
Esquizencefalia. Por defecto primario del desarrollo cerebral u oclusión de la arteria cerebral media.
Diagnóstico
Hidranencefalia. La ausencia de ecos en la zona anterior y media del cráneo la distingue de la hidrocefalia severa donde se ve una delgada línea de corteza cerebral y ecos de línea media. Este defecto es mortal dentro del primer año de vida.
Porencefalia. Quistes únicos o múltiples conectados con el sistema ventricular o espacio sub-aracnoideo ipsilateral. Estos no comprimen el cerebro y el hemisferio del lado dañado es usualmente más pequeño. El pronóstico depende de la extensión y localización del defecto.
Esquizencefalia. Hendidura que se extiende entre los ventrículos hasta el espacio sub-aracnoideo. Se asocia a ausencia de cavum de septo pellucidum. Son generalmente bilaterales y simétricos. El pronóstico es variable, pero la mayoría de los casos muestra retraso mental severo y convulsiones.
La diferencia entre la porencefalia y la esquisencefalia unilateral radica en que la porencefalia está delimitada por sustancia blanca y la esquisencefalia por sustancia gris. La diferenciación es poco importante desde el punto de vista pronóstico y es sólo posible mediante RNM 8.
Hernia diafragmática (HDC). Se presenta con frecuencia de 1 en 4000-10.000 embarazos. Se relaciona en un 30% con defectos anatómicos y alteraciones cromosómicas. Esta malformación se produce por defecto a nivel del diafragma y la herniación de parte del contenido abdominal hacia el tórax. El diagnóstico es difícil y en la mayoría de los fetos no se diagnostica en primer trimestre ya sea porque el defecto es pequeño o porque las vísceras abdominales aún no se han herniado por el defecto diafragmático (Figura 6).

Las hernias diafragmáticas pueden ser izquierdas (80%), derecha (17%) o bilateral (2%). Se clasifican en base a la relación que existe entre la circunferencia craneana y el área pulmonar del pulmón remanente (LHR, lung/head ratio), expresado en un porcentaje que compara esta medida con la esperable en un pulmón normal, o LHR O/E (lung/head ratio observado / esperado). Dependiendo de este porcentaje, estas se dividen en severa, extrema, moderada y leve. Esta clasificación está basada en la sobrevida que presentan en el periodo neonatal. Es así como las hernias diafragmática congénitas severas representan el 5% de las HDC y presentan una sobrevida menor al 5%. La extrema, corresponde al 10% de la HDC y presenta una sobrevida cercana al 15%. La moderada corresponde al 30% de las HDC y presenta una sobrevida del 50%. El resto de las HDC o leves presenta sobrevida mayor al 80%.
Las HDC clasificadas como extremas y severas son susceptibles a tratamiento intrauterino. El Grupo del Eurofetus (eurofetus.org) en trabajos randomizados ha mejorado la sobrevida de este grupo de HDC usando un balón de oclusión traqueal (FETO, fetal endoscopic tracheal occlusion) que insertado mediante fetoscopía a las 26 – 28 semanas y retirado a las 32 - 34 semanas logra aumentar los volúmenes pulmonares en base a la acumulación de secreciones pulmonares. La sobrevida con FETO es de 25% y 45% para la HDC extrema y severa.
Dentro de los criterios pronósticos de la hernia diafragmática se encuentra el LHR O/E, la presencia de hígado en tórax, siendo peor si este está en el tórax y la edad gestacional al nacimiento. La RMF es el examen con mayor sensibilidad para definir volumen pulmonar total y porcentaje de hígado en tórax, elementos claves para establecer el pronóstico de este defecto (23).
Alteraciones en extremidades (1 en 5.000- 1 en 10.000) Defectos como pie equino varo (Pie Bot), dedos sobrepuestos, polidactilia, alteraciones en la movilidad y ausencia de extremidades ser pueden diagnosticadas en primer trimestre y con mayor sensibilidad en 2do y 3er trimestre. Su importancia radica en la asociación de estos defectos con alteraciones cromosómicas. Es así como la polidactilia se asocia a trisomía 13, el equino varo y dedos sobrepuestos con trisomía 18 9.
Las alteraciones en la movilidad se asocian a la artrogriposis y se pueden diagnosticar al observar ausencia de movimientos fetales y una posición de flexión permanente durante el examen en un feto vivo.
Como parte de las alteraciones de las extremidades, las displasias esqueléticas constituyen un grupo importante dentro de las enfermedades raras. Estas se refieren a un espectro de alteraciones del crecimiento de los huesos, que comprende enfermedades letales y no letales. El diagnóstico prenatal de la variedad de displasia esquelética es dificultosa. Dado que el diagnóstico de ésta enfermedad implica alteración del crecimiento, la sospecha de éstas son escasas en la ecografía de primer trimestre y habitualmente la sospecha se inicia en el segundo trimestre con crecimiento bajo el percentil 5 de huesos largos (fémur-húmero-tibia- peroné-radio – cubito), signos de hipomineralización ósea, o la presencia de costillas anormales, por ejemplo.
Desde el punto de vista obstétrico es importante determinar si el defecto es letal (Osteogénesis Imperfecta tipo II, Displasia tanatofórica asfixiante, acondrogénesis). La definición de letal está dada principalmente por el desarrollo de hipoplasia pulmonar y tórax estrecho. La aproximación a la hipoplasia está dada por una circunferencia torácica bajo el percentil 5 para la curva gestacional, además de la presencia de polihidroamnios, la que es resultado de una mala mecánica ventilatoria fetal. Aquí cobra especial relevancia el uso de RMF para la estimación de volumen pulmonar total, medición que otorga una mayor sensibilidad para este diagnóstico.
Establecida la hipoplasia pulmonar como alteración asociada, la presencia de polihidroamnios y los elementos de hipomineralización son sugestivos de acondrogenesis, así como las costillas cortas lo son del síndrome de costilla corta letal o displasia torácica asfixiante.
Disgenesia Caudal (Síndrome de Body -Stalk). Se presenta en una frecuencia de 1 en 10.000 embarazos y se asocia en un 15% con alteraciones cromosómicas. El pronóstico es letal. Se diagnostica al observar un defecto abdominal mayor, xifo-escoliosis, arteria umbilical única con cordón corto y traslucencia nucal aumentada. Usualmente un extremo del feto se observa en la cavidad amniótica y otro extremo en la cavidad celómica, sugiriéndose que su origen sería una rotura localizado en el amnios 9.
V.- Aspectos generales del manejo en la Unidad de NeonatologíaComo equipo neonatal, el enfrentamiento de un paciente con sospecha de enfermedad rara requiere reunir todos los antecedentes entregados por el equipo obstétrico, con el fin de disponer, en el momento del nacimiento, de todo el personal, los equipos y medicamentos que sean necesarios para estabilizar al paciente y complementar el diagnóstico. Los primeros momentos luego del nacimiento son muy importantes; condiciones del recién nacido que requieran reanimación avanzada neonatal deben ser parte de las competencias tanto del personal médico como de las matronas de la unidad, para permitir la realización de maniobras en forma coordinada.
Según los antecedentes prenatales, se requerirá la presencia de distintos subespecialistas médicos en la unidad, como cirujano infantil, cardiólogo, genetista, radiólogo, neurólogo, con el objetivo de tener una valoración más completa del recién nacido. Si es necesario realizar algún procedimiento, como ecografía cerebral, ecocardiografía, electroencefalograma o alguna cirugía de urgencia, estos deben estar disponibles en forma temprana. La comunicación fluida con los servicios de apoyo, como radiología, laboratorio, farmacia y banco de sangre, permite completar la evaluación inmediata de estos pacientes. La solicitud precoz de exámenes sanguíneos, metabólicos o genéticos, (Cariograma, FISH, Screening Metabólico, por ejemplo) pueden hacer la diferencia en el resultado perinatal de estos pacientes.
El apoyo y la contención a los padres, requiere de conocimientos y destrezas comunes a todo el equipo de matronas y técnicos paramédicos, capacitados para enfrentarse a pacientes con requerimientos especiales desde sus primeras horas, entregando una atención integral que no tendrá restricción de horario y que favorecerá la progresiva construcción del vínculo padres-hijo. Es muy importante la realización de una visita prenatal de los padres a la Unidad de Neonatología, para transmitir aquella información que les permita enfrentar el momento del nacimiento de su hijo, los riesgos a los que lo expondrá su enfermedad y los procedimientos que se realizarán. Las reuniones periódicas de los padres con los múltiples especialistas, permiten el intercambio de información, la que debe ser clara y completa. Así, teniendo al neonatólogo como cabeza del equipo y con apoyo de psicólogos especialistas, deben crearse espacios donde se comenten los avances o cambios en la condición clínica y se resuelvan dudas durante la permanencia del recién nacido en la unidad.
ConclusiónLas enfermedades raras requieren un enfoque multidisciplinario desde la vida prenatal y a lo largo de su vida. El diagnóstico prenatal es la primera instancia para el diagnóstico y búsqueda de la etiología. Sin embargo, más importante que afinar el diagnóstico, situación que muchas veces se hace en el período postnatal, es desde el punto de vista médico el trabajo en equipo para así trazar un adecuado manejo obstétrico y un seguro paso a la vida extrauterina. De primordial importancia es orientar y educar a los padres sobre la enfermedad del hijo y prepararlos en los siguientes pasos a seguir.
El autor declara no tener conflictos de interés, en relación a este artículo.
Las imágenes de este articulo han sido autorizadas ‘por el paciente o sus padres para su publicación.
Referencia no citada[23].