







Las enfermedades autoinmunes son un grupo de enfermedades de relativo reciente conocimiento. Muchas de ellas están genéticamente determinadas (excepto el síndrome de PFAPA). Se caracterizan por episodios recurrentes de fiebre asociada a síntomas que generalmente pueden comprometer la piel, sistema músculo esquelético y gastrointestinal. A pesar de su baja prevalencia, el descubrimiento de los genes comprometidos en algunas de ella, ha permitido una mejor comprensión de los mecanismos de la respuesta inmune innata y en especial del rol de los llamados inflamosomas. Estos avances han permitido terapias más específicas, lo que ha llevado a disminuir en forma importante la morbilidad asociada, tanto a corto como a largo plazo. En el área pediátrica, el síndrome de PFAPA debe ser incluido como alternativa en el diagnóstico diferencial.
Autoimmune diseases are an emerging group of genetically determined diseases (except PFAPA) that affect innate immune system. They are characterized by recurrent episodes of fever associated with symptoms affecting skin, musculoskeletal and gastrointestinal system. Although unfrequent, the discovery of affected genes has allowed a better understanding of molecular mechanisms of innate immune response, specially about the role of inflammasomes. Subsequent targeted therapies have allowed a great improvement in short term and long term morbidity of most of these diseases. In children, PFAPA must be included in the analysis of differential diagnosis.
La fiebre es un síntoma muy frecuente en pediatría y causante de alarma en los padres. Generalmente las causas son cuadros virales autolimitados. Sin embargo, cuando el síntoma se hace frecuente y en especial si se repite, las alternativas etiológicas son de diagnóstico más complejo. Desde hace ya más de una década, se ha descrito un nuevo grupo de enfermedades: los síndromes autoinflamatorios. Estos comprenden un grupo creciente de desórdenes inflamatorios de carácter episódico, de baja prevalencia en general, cuya causa no se relaciona ni con infecciones, autoanticuerpos ni con linfocitos T (LT) autoreactivos. En la gran mayoría de ellos se ha podido establecer una alteración que involucra la repuesta inmune innata y muchas veces hereditaria.
Inicialmente se incluyeron 4 grupos de enfermedades, conocidas también como fiebres recurrentes hereditarias: las 3 primeras entidades o grupos son la fiebre mediterránea familiar (FMF), lejos la más frecuente dentro de este grupo de enfermedades, el síndrome periódico asociado al receptor de factor de necrosis tumoral (TNF) (TRAPS) y el grupo de las deficiencias de la mevalonato kinasa (MVK): aciduria mevalónica y el síndrome de hiper IgD (HIDS). El cuarto subgrupo, el de las enfermedades por criopirinas, incluye 3 entidades: el síndrome autoinflamatorio familiar por frío (FCAS), el síndrome de Muckle Wells (MWS) y la enfermedad inflamatoria multisistémica de inicio neonatal (NOMID o CINCA). Últimamente se ha agregado otro síndrome: el asociado a un dominio de oligomerización de nuclétido llamado NLRP12. Es curioso que la característica principal, de las 3 primeras entidades especialmente, es la aparición intermitente de síntomas clínicos que contrasta con la naturaleza genética y por tanto permanente, de sus defectos (1).
Cabe destacar la baja prevalencia en general de estas alteraciones. La más frecuente de todas es la fiebre mediterránea familiar en población con ascendencia turca, judía, armenia y árabe. En Turquía se ha descrito una prevalencia de 0,0027 a 0,82 % y en Arabia de 1/25. El TRAPS es la más frecuente dentro de las de herencia autosómica dominante: se han descrito algunas familias y aproximadamente 200 casos esporádicos. El grupo de las enfermedades por criopirinas alcanza alrededor de 200 casos reportados para cada una en el mundo.
A pesar de la baja frecuencia descrita, la intensidad y alta recurrencia de los síntomas afectan en forma muy importante la calidad de vida de los pacientes Por la misma razón de esta baja prevalencia, no es habitual su sospecha y frecuentemente los pacientes son sometidos a múltiples estudios para aproximarse al diagnóstico.
La presentación clínica común son episodios de crisis de inflamación intermitentes con manifestaciones generales y locales. Los síntomas generales son fiebre y elevación de reactantes de fase aguda. Los compromisos locales afectan principalmente 3 áreas: abdomen, piel y sistema músculo esquelético.
A lo largo de los años también se han incluido en este grupo de enfermedades los llamados desórdenes piogénicos hereditarios. Estos incluyen el síndrome de artritis piogénica, pioderma gangrenoso y acné (PAPA), el síndrome de Majeed (osteomielitis crónica multifocal recurrente y anemia) y el déficit de antagonista de receptor (R) de IL-1 (DIRA).
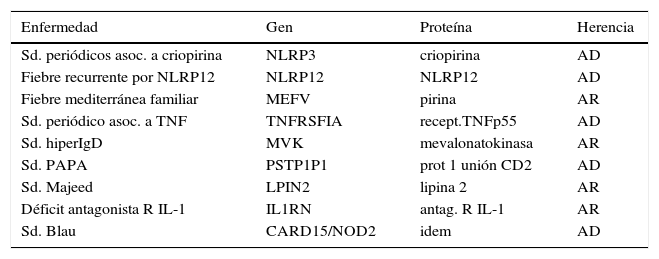
Clasificación de las enfermedades monogénicas autoinflamatorias
| Enfermedad | Gen | Proteína | Herencia |
|---|---|---|---|
| Sd. periódicos asoc. a criopirina | NLRP3 | criopirina | AD |
| Fiebre recurrente por NLRP12 | NLRP12 | NLRP12 | AD |
| Fiebre mediterránea familiar | MEFV | pirina | AR |
| Sd. periódico asoc. a TNF | TNFRSFIA | recept.TNFp55 | AD |
| Sd. hiperIgD | MVK | mevalonatokinasa | AR |
| Sd. PAPA | PSTP1P1 | prot 1 unión CD2 | AD |
| Sd. Majeed | LPIN2 | lipina 2 | AR |
| Déficit antagonista R IL-1 | IL1RN | antag. R IL-1 | AR |
| Sd. Blau | CARD15/NOD2 | idem | AD |
Traducido de: Rigante et al Theplodding diagnosis of monogenic autoinflammatory diseases in childhood: From clinical scenery to laboratory investigation. ClinChemLabMed 2011; 49(5): p784
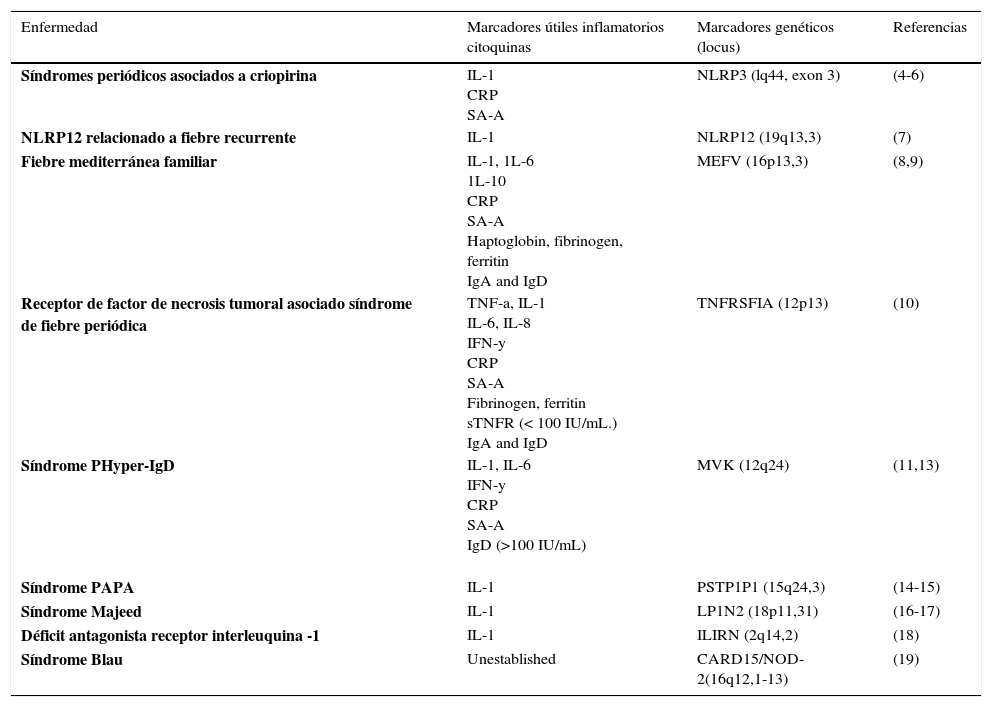
Marcadores de laboratorio y genéticos útiles para el diagnóstico de enfermedades autoinflamatorias en niños
| Enfermedad | Marcadores útiles inflamatorios citoquinas | Marcadores genéticos (locus) | Referencias |
|---|---|---|---|
| Síndromes periódicos asociados a criopirina | IL-1 CRP SA-A | NLRP3 (lq44, exon 3) | (4-6) |
| NLRP12 relacionado a fiebre recurrente | IL-1 | NLRP12 (19q13,3) | (7) |
| Fiebre mediterránea familiar | IL-1, 1L-6 1L-10 CRP SA-A Haptoglobin, fibrinogen, ferritin IgA and IgD | MEFV (16p13,3) | (8,9) |
| Receptor de factor de necrosis tumoral asociado síndrome de fiebre periódica | TNF-a, IL-1 IL-6, IL-8 IFN-y CRP SA-A Fibrinogen, ferritin sTNFR (< 100 IU/mL.) IgA and IgD | TNFRSFIA (12p13) | (10) |
| Síndrome PHyper-IgD | IL-1, IL-6 IFN-y CRP SA-A IgD (>100 IU/mL) | MVK (12q24) | (11,13) |
| Síndrome PAPA | IL-1 | PSTP1P1 (15q24,3) | (14-15) |
| Síndrome Majeed | IL-1 | LP1N2 (18p11,31) | (16-17) |
| Déficit antagonista receptor interleuquina -1 | IL-1 | ILIRN (2q14,2) | (18) |
| Síndrome Blau | Unestablished | CARD15/NOD-2(16q12,1-13) | (19) |
Traducido de: Rigante et al Theplodding diagnosis of monogenic autoinflammatory diseases in childhood : From clinical scenery to laboratory nvestigation ClinChemLabMed 2011;49(5): p784
Aún cuando se ha avanzado enormemente en la etiopatogenia de estos cuadros, su frecuencia es muy baja. Sin embargo, en pediatría existe un síndrome con características clínicas similares y con una prevalencia claramente mayor: el síndrome de fiebre periódica con adenitis, faringitis y aftas (PFAPA). Este cuadro no se hereda en forma mendeliana, es de buen pronóstico y debe constituir siempre parte del diagnóstico diferencial.
Finalmente, se ha propuesto expandir el concepto de autoinflamación a otras enfermedades inflamatorias sin una base genética tan clara, pero con alteración de la respuesta inmune innata. Entre ellas: artritis idiopática juvenil (AIJ), enfermedad de Still del adulto e incluso la gota. En las dos primeras se ha observado una notable respuesta a anakinra y también se ha descrito una marcada elevación de la IL-18, lo que podría contribuir a esclarecer su etiopatogenia a futuro (2).
Probablemente muchas de las enfermedades inflamatorias no clasificables sin compromiso de la respuesta adaptativa son también enfermedades del sistema inmune innato. Así, las enfermedades inflamatorias podrían ser parte de un continuo entre dos polos: desde la autoinmunidad a la autoinflamación.
Los estudios a nivel de ciencia básica han logrado establecer que en la mayoría de estas enfermedades, la alteración fisiopatológica común parece ser la sobre estimulación de las señales que favorecen la producción de IL-1.
En forma muy amplia y general, el sistema inmunológico es capaz de reconocer “lo propio” y desconocer “lo extraño”.
En la respuesta adaptativa, se estimulan receptores de células T y B, que sufren mutaciones somáticas y llevan finalmente a una fina especificidad del receptor frente al reencuentro con el antígeno correspondiente. Esto produce un amplio repertorio de receptores y permite el desarrollo de la memoria inmunológica.
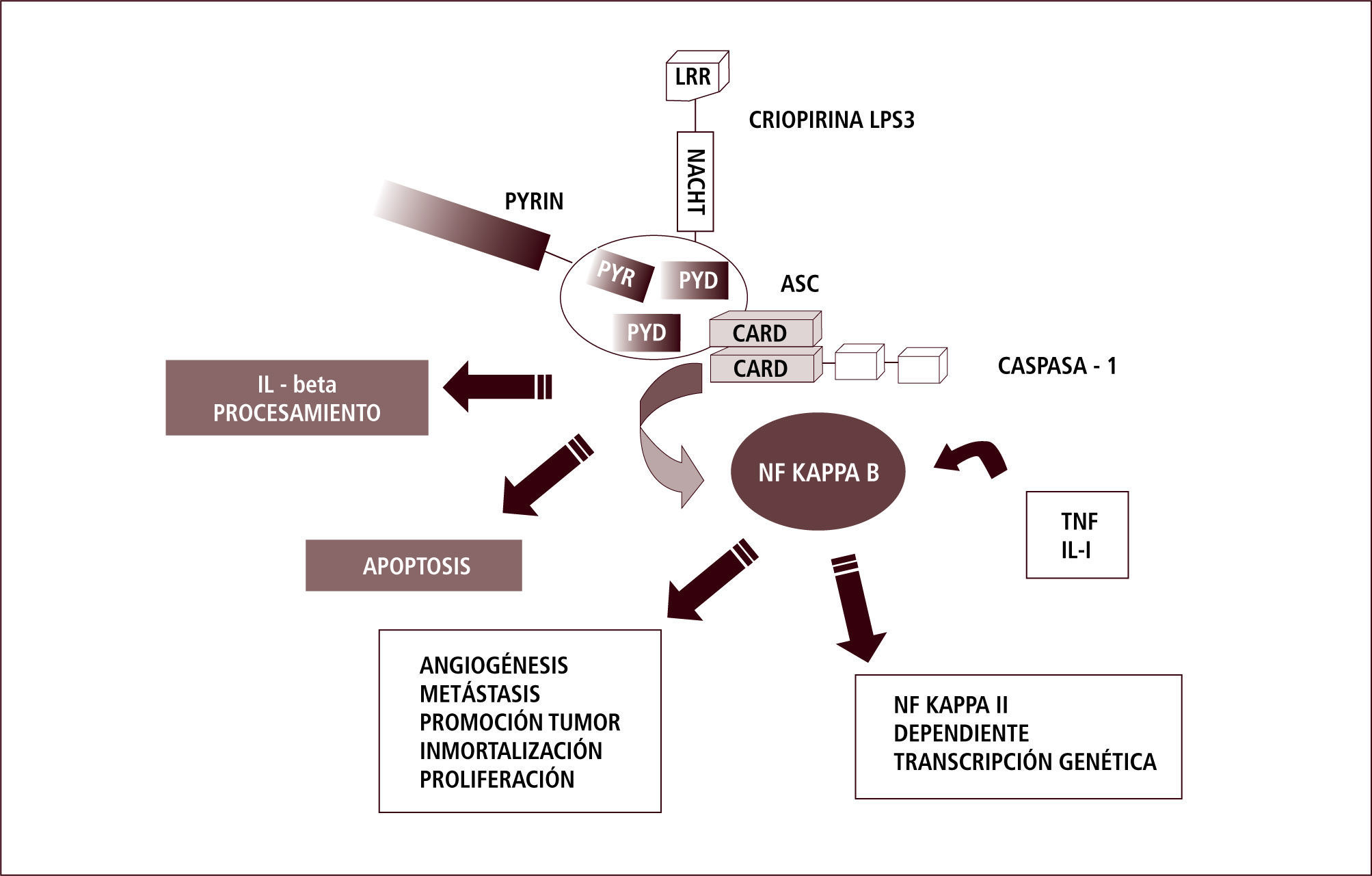
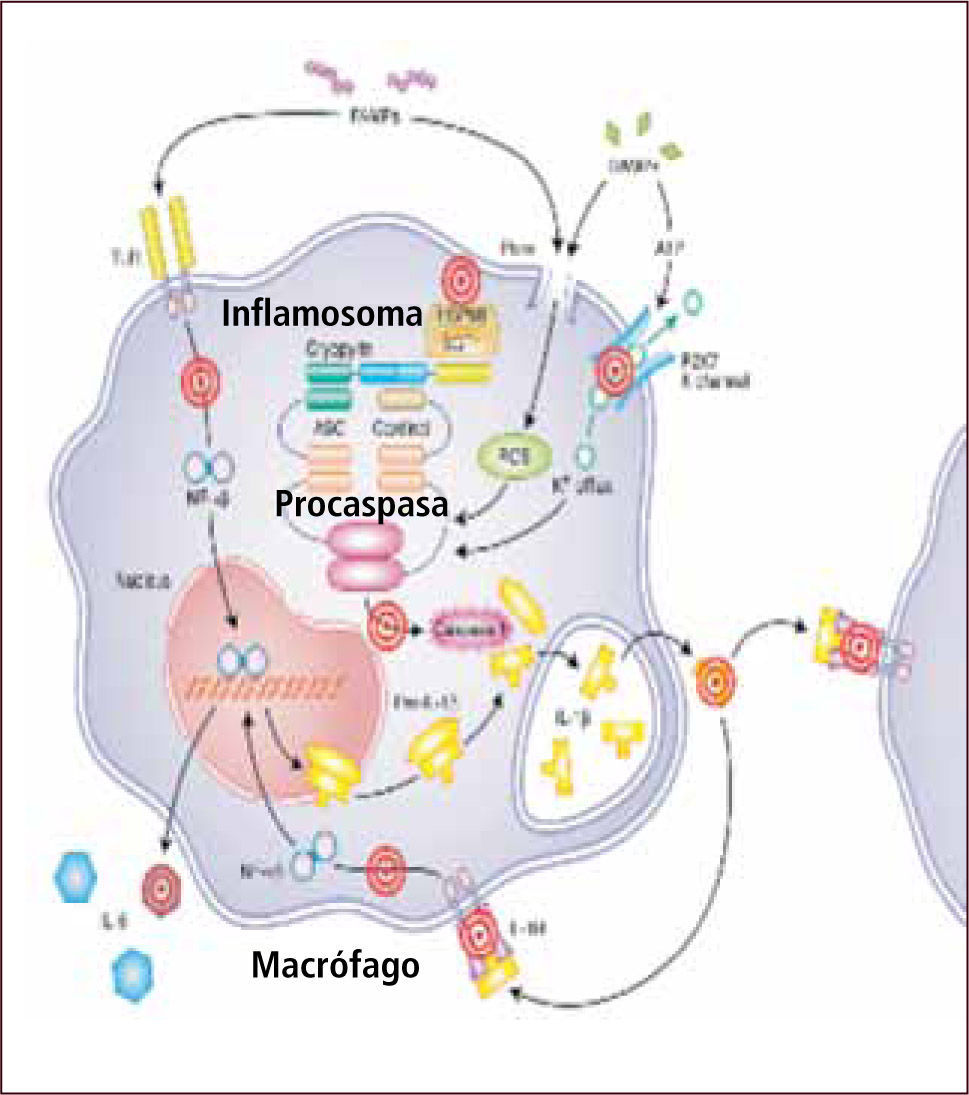
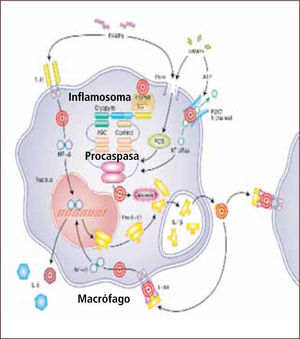
En la respuesta innata, el descubrimiento de los receptores tipo Toll (TLR) nos ha permitido comenzar a entender cómo esta primera vía es capaz de distinguir lo propio de lo extraño. Los TLR están localizados en la membrana y pueden “sensar” o reconocer estructuras particulares, conservadas en los patógenos a lo largo del tiempo, denominadas PAMPs (patrones moleculares asociados a patógenos). Posteriormente se han descubierto “sensores” intracitoplasmáticos como contraparte de los TLRs. Entre estos, se encuentra una familia de proteínas: los receptores tipo NOD (NLR). Estos son capaces de reconocer otro tipo de señales: aquellas asociadas a daño o stress celular. Estos patrones moleculares se denominan DAMPs (patrones moleculares asociados a daño). Ambos receptores son instrumentos para iniciar la respuesta inflamatoria, que a su vez gatilla la respuesta inmune innata y adaptativa. Algunos de estos receptores intracelulares van a llevar a la formación de los llamados inflamosomas: estructuras moleculares que llevan a estimulación y secreción de citoquinas inflamatorias, especialmente IL-1 beta (3).
La IL-1 beta es una citoquina inflamatoria muy potente. Su actividad está determinada por el clivaje proteolítico de su precursor inactivo. Este clivaje es producido por la caspasa-1, que a su vez es activada dentro de estos complejos proteicos intracelulares denominados “inflamosomas” (4).
En forma más precisa, la familia de los receptores tipo NOD (NLR) tiene homología estructural con proteínas de resistencia vegetal (involucradas en la interacción huésped-parásito). Esta familia de receptores comprende 14 receptores NALP y 5 NOD. Estos receptores forman complejos multimoleculares llamados “inflamosomas”. El más estudiado es el NALP3 que se relaciona con el aumento, vía caspasa 1, del procesamiento y secreción de IL-1 beta. Las pirinas y criopirinas son parte de este grupo de “sensores” de daño intracelular Así, la presencia de defectos funcionales en sus genes o dominios se han relacionado con defectos en las proteínas apoptóticas y en señales de transducción (5, 6).
La mayor parte de los pacientes que presenta fiebres periódicas hereditarias tiene mutaciones, ya sea en las pirinas o en el receptor de TNF, ambos íntimamente involucrados en la respuesta inmune innata. En forma muy general, tanto las pirinas como las criopirinas parecen modular la actividad de proteínas apoptóticas y de vías de transducción de señales y juegan, por lo tanto, un rol muy importante en las vías de infamación del sistema inmune innato (7).
EpidemiologíaEn general las enfermedades auto-inflamatorias son entidades poco frecuentes y su incidencia depende de la distribución del gen involucrado en determinadas poblaciones. La enfermedad autosómica recesiva más frecuente de este nuevo grupo es la FMF (hasta 1 en 25), y predomina en población del área mediterránea oriental (turcos, judíos no Ashkena-zi, árabes y armenios). Entre aquellas con herencia autosómica dominante, la más frecuente es TRAPS (8).
Enfrentamiento general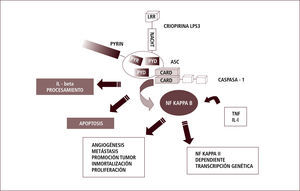
El diagnóstico de las fiebres periódicas recurrentes constituye un desafío. Como siempre, debe hacerse hincapié en una buena historia clínica, antecedentes familiares y étnicos, registro de los episodios, y examen físico (10-17). Varias condiciones clínicas pueden aparecer ini-cialmente como enfermedades autoinfamatorias: infecciones ocultas o recurrentes, inmunodefciencias, enfermedades autoinmunes atípicas o enfermedades malignas (18, 19). También es frecuente la sobreposición de síntomas y signos, siendo a veces muy difícil la clasificación en los distintos grupos descritos hasta ahora. Muchos pacientes con alta sospecha clínica, pueden no cumplir los criterios para incluirlos en las enfermedades actualmente descritas. La comprensión de estos cuadros “indiferenciados” probablemente mejorará en futuro cercano con los avances en investigación en el área.
Es frecuente observar durante las crisis: fiebre, artritis/artralgia, compromiso del estado general, dolor abdominal y rash cutáneo. Habitualmente se inicia en edad infantil o adolescencia. La regularidad de las crisis es muy variable. En los períodos intercrisis, los pacientes habitualmente son asintomáticos, pero pueden existir inflamación subclínica. La complicación a largo plazo mas importante y temida es el desarrollo de amiloidosis.
En general el diagnóstico se basa en la clínica y los antecedentes étnicos. Particularmente, en el caso de MKD, pueden realizarse mediciones enzimáticas. La realización de exámenes genéticos debe orientarse según los hallazgos clínicos.
A continuación describiremos la presentación clínica y el manejo recomendado para los cuadros clínicos más conocidos hasta ahora.
Fiebre mediterránea familiar (FMF)Es una enfermedad autosómica recesiva que afecta primordialmente grupos étnicos del área del Mediterráneo. Se describió como entidad en 1945.
El gen responsable, MEFV, se encuentra en el brazo corto del cromosoma 16. Este gen codifica para una proteína: pirina/marenostrin, ubicada casi exclusivamente en los neutrófilos y sus precursores. Se cree que su rol es disminuir la inflamación a nivel de estas células.
El hecho clínico característico son los episodios de fiebre y serositis con inflamación aguda importante. Se inicia habitualmente antes de los cinco años. En poblaciones con alta prevalencia, pueden usarse los criterios de Tel Ashomer. En caso contrario o en presentaciones atípicas, podría hacerse estudio genético.
Las crisis se inician con fiebre alta, duran entre horas 3 a 4 días. La frecuencia es muy amplia y puede variar entre semanal a una crisis cada varios años. Existe respuesta inflamatoria en las crisis y en los intervalos también puede objetivarse inflamación por exámenes de laboratorio. Se desconocen los gatillantes específicos, pero se describen el ejercicio intenso, stress emocional o ciclos hormonales.
Las serositis más frecuentes son: peritonitis (90%) y pleuritis (45%). Puede confundirse con apendicitis. El compromiso de piel se manifiesta en lesiones tipo erisipela, habitualmente en relación con artritis de extremidades inferiores. La sinovitis habitualmente compromete pocas articulaciones y no deja secuelas articulares. Otras manifestaciones menos frecuentes son vasculitis, orquitis, meningitis aséptica y mialgias. El hallazgo de proteinuria es sugerente de amiloidosis (que es la complicación más importante y potencialmente letal).
Criterios diagnósticos de Tel-Ashomer para el diagnóstico de FMFCriterios mayores- 1.
Episodios de fiebre recurrente con peritonitis, pleuritis o sinovitis
- 2.
Amiloidosis de tipo AA (sin factores predisponentes)
- 3.
Respuesta favorable a dosis de colchicina diaria
- 1.
Episodios febriles recurrentes
- 2.
Eritema tipo erisipela
- 3.
Historia familiar de FMF en pariente de 1 grado
Diagnóstico definitivo: 2 criterios mayores o 1 mayor y 2 menores
probable: 1 criterio mayor y 1 menor
El tratamiento de esta entidad es la colchicina. Su uso diario previene las recurrencias y el desarrollo de amiloidosis. La dosis habitual es 1mg/día, pero puede aumentarse hasta 2,5mg/día. En general es una droga bastante segura, pero se ha reportado toxicidad severa cuando se usa en edades mayores, falla hepática o renal y uso concomitante de macrólidos. En el pequeño grupo de pacientes no respondedores a colchicina (5 a 10%) se ha propuesto el uso de anakinra (antagonista recombinante del receptor de IL-1) con buenos resultados.
Síndrome periódico asociado a receptor de tnf (TRAPS)Este síndrome se describió por 1 vez en 1982 en pacientes de ascendencia irlandesa o escocesa y fue la primera de las enfermedades auto-inflamatorias en que se describió la mutación genética específica (1999). Es un cuadro autosómico dominante de inicio habitualmente en edad preescolar. Se caracteriza por fiebre recurrente, dolor abdominal, mialgias migratorias, artralgias/artritis, pleuritis, rash, conjuntivitis o edema periorbitario. Las lesiones de piel pueden ser tipo placas urticariales, centrífugas y sensibles. Las mialgias son muy sugerentes de TRAPS y se deben probablemente a una fasceítis monocítica.
Las crisis febriles suelen ser prolongadas y se presentan con una frecuencia de entre 2 a 6 veces al año.
El laboratorio muestra leucocitosis, reactantes de fase aguda elevados y elevación de inmunoglobulinas (especialmente IgA). Estas alteraciones también pueden objetivarse en períodos intercrisis. En general el pronóstico es benigno, pero se ha reportado desarrollo posterior de amiloidosis entre un 10 a un 25%, por lo que debe estudiarse siempre esta posibilidad.
Existen propuestas de criterios diagnósticos, pero no han demostrado gran especificidad. Así, el hallazgo más discriminatorio es una baja concentración del TNFR p55 soluble (< 1 ng/ml) en períodos intercrisis. Por el momento debe realizarse un estudio genético que compruebe mutaciones en el gen TNFRSF1A. Se han involucrado varias mutaciones (más de 82) en la línea germinal del gen TNFRSF1A (superfamilia del 1 A), localizado en el cromosoma 12. Este gen se expresa en varias células y se la relaciona con inducción de secreción de citoquinas, expresión de moléculas de adhesión leucocitarias, resistencia a patógenos intracelulares, pirexia y caquexia.
En estos pacientes, la colchicina no previene la recurrencia de los ataques. El uso de corticoides desde el inicio de las crisis puede atenuar la extensión y severidad de las mismas. Dada la evidencia del rol del TNF en las crisis, se planteó el uso de sus inhibidores, como el etanercept. Algunos pacientes responden a su uso, logrando disminuir la frecuencia de las crisis y disminuir las dosis de corticoides requeridas. También se ha probado infliximab y anakinra, con resultados variables.
Síndrome de hiper igd (HIDS)HIDS es un cuadro de herencia autosómica dominante, causado por mutaciones en el gen MVK (localizado en 12q24). El resultado de la mutación es una función alterada de la mevalonato kinasa, que es una enzima del peroxisoma involucrado en las vías de metabolización del colesterol. Se han reportado más de 100 mutaciones, siendo la más frecuente V377I que se asocia con una clínica más leve por una función residual de la enzima.
Es muy frecuente en población holandesa, por lo que también se conoce como “fiebre periódica de tipo holandés”, pero existen reportes en otras razas.
Habitualmente se inicia en la infancia precoz. Las crisis duran entre 3 a 7 días, muchas veces gatilladas por vacunaciones, infecciones bacterianas, stress emocional o menstruación. Los síntomas son fiebre alta, cefalea, adenopatías cervicales, esplenomegalia, dolor abdominal intenso, diarrea o vómitos, artralgias, rash polimorfo, úlceras orales o vaginales. Tiene menos riesgo de desarrollar amiloidosis que otras EAI.
El laboratorio muestra elevación de reactantes de fase aguda en forma intensa y moderado consumo de complemento. Es característico el ascenso de niveles de IgD (>100UI/ml) tanto en las crisis como en los períodos intercrisis. Esta elevación policlonal de IgD no es exclusiva de HIDS (también en FMF y TRAPS), pero en estos otros cuadros habitualmente no alcanza niveles tan altos. Existe aproximadamente un 20% de pacientes que no tienen IgD aumentada. El hallazgo específico es el aumento en la excreción urinaria de ácido mevalónico durante las crisis.
Debe distinguirse el HIDS de la aciduria mevalónica, con excreción aumentada de ácido mevalónico crónicamente. Estos pacientes tienen deficiencia absoluta de la enzima, y cursan con retardo mental, microcefalia, atrofia cerebelar, distrofia retinal, dismorfias y fiebre periódica.
El tratamiento habitualmente incluye el uso de AINEs para las crisis. Eventualmente, algunos pacientes responden a corticoides. La colchicina no constituye una alternativa eficaz. Dado que su etiopatogenia implica una acumulación de mevalonato, se ha planteado el uso de simvastatina, con resultados variables. Por ahora no existe un tratamiento óptimo.
Síndromes periódicos asociados a la criopirinaConstituyen un subgrupo de las enfermedades autoinflamatorias, que comprende 3 entidades de herencia autosómica dominante. Estas son: el síndrome autoinflamatorio familiar por frío (FCAS), síndrome de Muckle Wells (MWS) y el síndrome infantil crónico neurológico, cutáneo y articular (CINCA o NOMID). Se ha planteado que estas alteraciones son variantes alélicas de un espectro, que va desde la más leve a la más severa en el orden anteriormente expuesto.
Las manifestaciones clínicas de inflamación están causadas por una activación constitutiva del inflamosoma debido a criopirinas mutantes, lo que resulta en una liberación persistente de IL-1 activa. La mayoría presentan alteraciones en el dominio NACHT del gen NLRP3 y son ejemplos de desregulaciones en el procesamiento y secreción de IL-1.
Habitualmente se inician durante la infancia, con lesiones en piel tipo urticaria (pero que en la biopsia tienen predominio de infiltrado neutrofílico/linfocítico y no de mastocitos como se observa en las urticarias reales). FCAS y MWS suelen tener clínica sobrepuesta, con fiebre, rash, inflamación ocular y articular. CINCA o NOMID en cambio, se inicia en el período neonatal con rash persistente, artropatía hipertrófica generalmente de rodilla, meningitis crónica aséptica, ventriculomegalia, papiledema y sordera. Existe riesgo de amilodosis especialmente en CINCA y MWS (20 a 25%).
El laboratorio muestra elevación de reactantes de fase aguda, especialmente en el cuadro más severo (CINCA). Sin embargo es necesario el estudio genético para corroborar el diagnóstico, aun cuando sólo el 60% de los pacientes con CINCA tiene mutaciones específicas del NLRP3. De hecho, SEN el 2008 se describió otra entidad muy similar en clínica con FCAS (fiebre recurrente, síntomas articulares y rash gatillado por frío) que tiene una mutación en el NLRP12. Este gen está involucrado en el reconocimiento de moléculas microbiológicas y tiene también un rol en la activación del inflamosoma.
El tratamiento de elección en estos síndromes apunta al bloqueo de la IL-1. El uso de anakinra (antagonista recombinante del receptor de IL-1) ha demostrado ser muy exitoso en estos pacientes. Este medicamento logra tanto frenar las crisis de inflamación en forma muy rápida, como también ha probado ser efectivo incluso en las manifestaciones del SNC del NOMID.
Desórdenes hereditarios piogénicosSe incluyen en este grupo el síndrome de PAPA, el síndrome de Majeed y la deficiencia del antagonista del receptor de IL-1. La característica común es el compromiso óseo inflamatorio recurrente.
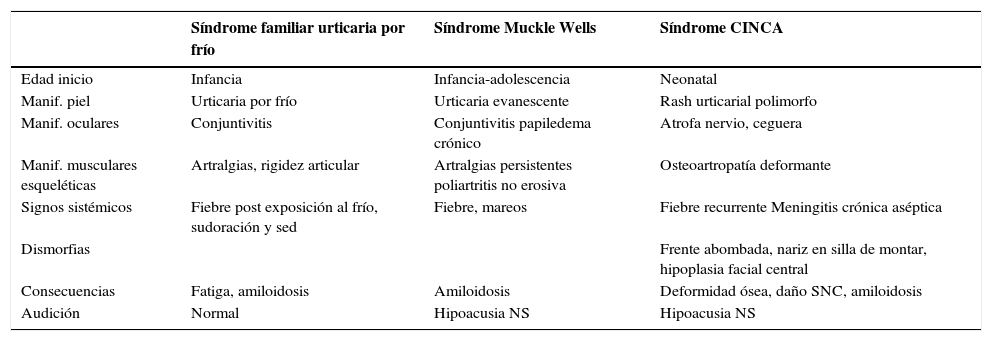
Clínica general de síndromes periódicos asociados a criopirinas
| Síndrome familiar urticaria por frío | Síndrome Muckle Wells | Síndrome CINCA | |
|---|---|---|---|
| Edad inicio | Infancia | Infancia-adolescencia | Neonatal |
| Manif. piel | Urticaria por frío | Urticaria evanescente | Rash urticarial polimorfo |
| Manif. oculares | Conjuntivitis | Conjuntivitis papiledema crónico | Atrofa nervio, ceguera |
| Manif. musculares esqueléticas | Artralgias, rigidez articular | Artralgias persistentes poliartritis no erosiva | Osteoartropatía deformante |
| Signos sistémicos | Fiebre post exposición al frío, sudoración y sed | Fiebre, mareos | Fiebre recurrente Meningitis crónica aséptica |
| Dismorfias | Frente abombada, nariz en silla de montar, hipoplasia facial central | ||
| Consecuencias | Fatiga, amiloidosis | Amiloidosis | Deformidad ósea, daño SNC, amiloidosis |
| Audición | Normal | Hipoacusia NS | Hipoacusia NS |
Traducido de: Rigante et al The plodding diagnosis of monogenic autoinflammatory diseases in childhood: From clinical scenery to laboratory investigation ClinChemLabMed 2011;49(5): p785
En el PAPA, las mutaciones comprometen el gen PSTPIP1, que afecta la unión de una proteína involucrada en la reorganización de la actina y por tanto en la formación del citoesqueleto. Esta proteína interactúa con la pirina, llevando a una apoptosis disregulada. La clínica se presenta con oligoartritis autolimitadas (con acumulación de material estéril sinovial rico en neutrófilos) y lesiones cutáneas purulentas desfigurantes. Se ha usado corticoides, inhibidores de TNF y últimamente, Anakinra.
El síndrome de Majeed se caracteriza por fiebre recurrente, osteomielitis recurrentes de inicio precoz, lesiones de piel inflamatorias transitorias y anemia diseritropoyética congénita. Se ha involucrado el compromiso del gen LPIN2. El tratamiento usado hasta ahora para el compromiso óseo y cutáneo comprende corticoides, AINEs, terapia física y transfusiones.
En el 2009 se describieron los primeros casos de déficit de antagonista del receptor IL-1. Son pacientes recién nacidos, con erupción pustular extensa, artritis y osteomielitis multifocal estéril. La alteración involucra mutaciones en el gen IL1RN del cromosoma 2. El antagonista del R de IL-1 inhibe su actividad y por tanto modula las respuestas inflamatorias e inmunes. Dada su etiopatogenia, esta entidad puede ser tratada con anakinra.
El diagnóstico de estas entidades se basa en la clínica y radiología, pero deben efectuarse los tests genéticos correspondientes para precisarlas.
Clínica de los desórdenes biogénicos infantiles hereditarios
| Síndrome de PAPA | Oligoartritis piogénica estéril, pioderma gangrenoso, acné quístico |
| Síndrome de Majeed | Osteomielitis recurrente multifocal de inicio precoz, anemia macrocítica hipocroma congénita, dermatosis inflamatoria transitoria |
| Déficit antagonista R IL-1 | Erupción cutánea pustular, osteomielitis multifocal Estéril de inicio neonatal |
Traducido de: Rigante et al Theplodding diagnosis ofmonogenicautoinflammatorydiseases in childhood: from clinical scenery to laboratory investigation ClinChemLabMed 2011;49(5): p788.
El uso de corticoides sistémicos y locales ha sido efectivo para el manejo de las manifestaciones articulares y cutáneas del PAPA, pero los pacientes pueden hacerse refractarios a ellos en el tiempo. Existen reportes muy auspiciosos con el uso de anakinra.
En el caso del síndrome de Majeed, las alternativas terapéuticas son amplias e incluyen el uso de AINEs, corticoides, interferon alfa o gamma, azitromicina, bifosfonatos e inhibidores de TNF.
Artritis granulomatosasActualmente se denomina como artritis granulomatosas pediátricas al grupo de enfermedades constituidas por el síndrome de Blau (forma familiar) y la sarcoidosis de inicio precoz (forma esporádica).
El síndrome de Blau es una enfermedad rara de herencia autosómica dominante. Afecta a niños pequeños de ambos sexos sin predilección de raza. Se caracteriza por poliartritis recurrentes granulomatosas, uveítis (con riesgo de cataratas y glaucoma) y rash ictiosiforme. La histología muestra granulomas no caseosos y las articulaciones pueden verse muy aumentadas de volumen en el tiempo.
La alteración genética ocurre a nivel del gen CARD15/NOD-2, descubierto el 2001. Este gen es parte de la familia de los receptores tipo NOD (proteínas intracelulares), que se expresan en monocitos y condor-citos. El síndrome de Blau presenta una mutación en el dominio NACHT del gen CARD15/NOD-2, lo que produce un aumento de la actividad de del NF kappa beta. Algunas variantes en el dominio LRR del mismo gen se han asociado con un subgrupo de pacientes en enfermedad de Crohn, enfermedad granulomatosa que afecta el tracto gastrointestinal. Por ello, algunos han planteado que debiera incluirse esta entidad en el grupo de las enfermedades autoinflamatorias. En el síndrome de Blau, no existe compromiso de vías aéreas y esto lo diferencia de la sarcoidosis de inicio precoz, que es la forma de presentación esporádica.
El diagnóstico del síndrome de Blau se basa tanto en la clínica como en la determinación del gen involucrado. El tratamiento comprende el uso de corticoides, drogas inmunosupresoras (Metotrexato y Ciclosporina), Anakinra e Infliximab (anti TNF).
Los pacientes con manifestaciones más leves, responden a AINEs, pero en el caso de cuadros más severos, se plantea el uso de corticoides sistémicos. Pueden usarse también drogas inmunosupresoras como MTX, ciclosporina y etanercept. El uso de anakinra en algunos reportes también ha mostrado buenos resultados.
Síndrome de fiebre periódica con aftas, faringitis y adenitis cervical (PFAPA)Es una enfermedad de causa desconocida (esporádica, no genética hasta ahora), descrita ya en 1987que se caracteriza por fiebre periódica, asociada a faringitis, adenitis cervical y aftas. Puede acompañarse de cefalea, dolor abdominal y/o artralgias.
Las crisis ocurren cada 4 a 6 semanas, duran 4 a 5 días y ceden espontáneamente. La fiebre es alta (hasta 40ºC), con mucho compromiso del estado general, pero los episodios no dejan secuelas. Puede iniciarse precozmente (alrededor de los 4 años), pero últimamente se han descrito también casos en adultos. El compromiso faríngeo simula en todo una faringitis estreptocócica, pero los cultivos son persistentemente negativos. La ausencia de aftas no descarta el síndrome (en las últimas series las describen sólo en un 22%).
Los niños afectados tienen buen desarrollo pondoestatural, no presentan otras enfermedades habitualmente ni tienen secuelas.
Diferencias en los criterios diagnósticos entre dos centros para el diagnóstico de pfapa
| TOMAS ET AL Fiebre recurrente regularmente de inicio precoz (antes de los 5 años) Síntomas sistémicos en ausencia de infección respiratoria alta, con al menos 1 de los siguientes signos :
|
| PADEH ET AL Fiebre mensual o cíclica en cualquier grupo etario
|
Traducido de: Padeh S et al Auto-inflammatory fever síndromes et al RheumDisClin N Am 2007; 33: p 601.
La causa del PFAPA es aún desconocida. Destaca la gran similitud con los episodios de neutropenia cíclica no complicados, por lo que se ha planteado que pudieran compartir vías comunes de desregulación. Estudios en pacientes con PFAPA en crisis muestran elevación de INF gamma, TNF e IL-6. Se ha planteado que quizás sea una repuesta inmune anormal frente a microorganismos comensales de las amígdalas o la mucosa oral.
La respuesta inmediata frente a una dosis de corticoides sugiere más bien una causa relacionada con la secreción de citoquinas inflamatorias más que con una infección. Además, la ausencia de casos secundarios en parientes o contactos cercanos, la falta de concentración de casos en forma estacional o geográfica y la persistencia sin progresión por largo tiempo, también hacen poco probable una causa infecciosa.
Laboratorio- -
Hemograma: sin anemia, leucocitosis leve
- -
VHS moderadamente elevada (40-50), plaquetas normales (intercrisis: VHS normal)
- -
IgD habitualmente normal o moderadamente elevada (menor 100)
- -
IgE puede ser elevada
- -
Serología autoinmune (-)
- -
Subpoblaciones de LT normales
- -
Imágenes en búsqueda de foco infeccioso (-)
Debe hacerse, entre otros, con enfermedad de Crohn (podría ser una manifestación precoz y luego aparecer síntomas GI), inmunodeficiencias (incluidas las neutropenias cíclicas y VIH), síndrome de Behcet y HIDS.
TratamientoLos glucocorticoides son característicamente, muy efectivos en yugular los síntomas.
Se recomienda el esquema con:
Prednisona 2mg/kg/dosis o betametasona 0,3mg/kg/dosis.
Puede usarse 1 dosis al inicio, otra a la mañana siguiente y la mitad en el día 3 y 4.
Con la primera dosis ya se observa respuesta de la fiebre a las 2 a 4 hrs. Las aftas pueden demorar más en responder. Con el tiempo, puede lograrse el mismo efecto con dosis menores. Los corticoides no previenen crisis a futuro, pero a largo plazo puede observarse espaciamiento de ellas (20).
El síndrome tiende a desaparecer en 6 a 10 años promedio.
Otra alternativa que se ha planteado es la tonsilectomía, con o sin adenoidectomía. Diversos estudios han sido poco claros en demostrar su beneficio. Revisiones recientes con metodología de metanálisis o Cochrane son también discordantes en sus conclusiones: el meta análisis del 2010 plantea la opción quirúrgica como la más efectiva a largo plazo, en cambio la revisión Cochrane de enero del mismo año plantea que debe sopesarse el riesgo de la cirugía con el manejo médico que mas bien aborta las crisis (y en el largo plazo eventualmente las distancia) (20, 21). En nuestro país, el grupo de la Universidad Católica publicó un reporte descriptivo de casos (22). Un estudio reciente del mismo grupo (presentado como poster en el Congreso de la Sociedad Chilena de Pediatría 2011) siguió a 8 pacientes con PFAPA. A todos se les realizó amigdalectomía. Ninguno presentó recurrencia de las crisis febriles, adenitis ni faringitis, pero sin embargo 25% presentó recurrencia de las estomatitis.
Claves para el diagnóstico de eai
Detalles sugerentes de EAI
|
Factores diferenciadores entre EAI
|
Traducido de: Rigante et al Theplodding diagnosis ofmonogenicautoinflammatorydiseases in childhood: fromclinicalscenerytolaboratoryinvestigation ClinChemLabMed 2011;49(5): p788
Claves específicas para el diagnóstico de eai
| Enfermedad específca | Clínica |
|---|---|
| Sd. periódicos asociados a criopirina | Rash tipo urticaria, hipoacusia neurosensorial, compromiso osteoarticular variable, periodicidad circadiana de los síntomas |
| Fiebre mediterránea familiar | Eritema tipo erisipela en pie o tobillo, respuesta a colchicina |
| TRAPS | Crisis febriles de larga duración, con mialgias migratorias, edema periorbitario con conjuntivitis, respuesta a corticoides |
| HIDS | Crisis febriles de inicio el 1 año (generalmente gatilladas por vacunaciones) más signos cutáneos, intestinales, linfadenopatía y esplenomegalia |
| Desórdenes piogénicos hereditarios | Manifestaciones agudas biogénicas de hueso, articulaciones y piel |
| Síndrome de Blau | poliartritis granulomatosa, uveítis y rash ictiosiforme |
Traducido de: Rigante et al Theplodding diagnosis ofmonogenicautoinflammatorydiseases in childhood: fromclinicalscenerytolaboratoryinvestigation ClinChemLabMed 2011;49(5): p789
Las enfermedades autoinflamatorias son un grupo de patologías, frecuentemente hereditarias, que afectan principalmente el sistema inmune innato. Se manifiestan generalmente por episodios de fiebre recurrente asociados a compromiso de piel, sistema músculo-esquelético y aparato gastrointestinal. A pesar de su escasa frecuencia, el descubrimiento de los genes involucrados en algunas de ellas ha aportado un gran conocimiento en relación a los mecanismos involucrados en la respuesta inmune primaria, especialmente en relación a los denominados “inflamosomas”, y, por otro lado, ha favorecido el desarrollo de nuevas terapias específicas, con lo que se ha podido mejorar la calidad de vida de estos pacientes. El diagnóstico debe basarse en los hallazgos clínicos y exámenes de laboratorio en el tiempo, teniendo siempre presente el diagnóstico diferencial de patologías especialmente infecciosas, autoinmunes y oncológicas. En pediatría, debe descartarse según la clínica, el síndrome de PFAPA. El factor genético es de crucial importancia, pero su estudio debe ser dirigido y correlacionado con las alteraciones inmunológicas asociadas.
La autora declara no tener conflictos de interés, con relación a este artículo.