




Aunque pueda parecer paradójico, las inmunodeficiencias primarias y la secundaria a infección por VIH frecuentemente se complican con enfermedades autoinmunes. Esto debido a la desregulación del sistema inmune y a la activación policlonal debida a infecciones recurrentes.
Se revisan diversas enfermedades autoinmunes y autoanticuerpos asociados con ambos tipos de inmunodeficiencias.
Las enfermedades autoinmunes pueden ser la primera manifestación de una inmunodeficiencia, por lo que deben estudiarse especialmente si la enfermedad autoinmune es atípica. Las patologías más frecuentemente asociadas son las citopenias autoinmunes y los enfermedades reumatológicas.
Debe realizarse una exclusión completa de las infecciones coincidentes o posiblemente causantes de complicaciones autoinmunes antes de iniciar tratamientos específicos para ellas.
Although it may seem paradoxical, primary immunodeficiencies and HIV immunodeficiency are frecuently complicated by autoimmune conditions.
This is because of the immune system disregulation and polyclonal activation due to recurrent infections.
We review various autoimmune diseases and autoantibodies associated with both types of immunodeficiencies.
Autoimmune diseases my be the first manifestation of an immunodeficiency, so we should screen for it, specially if this autoimmune disease is atypical. The most frecuent disease associated with immunodeficiencies are autoimmune cytopenias and rheumatologic disorders.
A through exclusion of infections coincident with or possibly causative of autoimmune complication should be undertaken before initiating specific treatments for autoimmune disease in this patients.
Los manifestaciones de autoinmunidad tanto clínicas como de laboratorio son hallazgos frecuentes en las inmunodeficiencias, a pesar de que pudiera pensarse que esto un hecho paradójico, sobre todo en aquellas inmunodeficiencias en las cuales hay una disminución en la producción de anticuerpos. Sin embargo, debido a que la desregulación inmunológica es un hecho frecuente en estos casos, la autorreactividad no es infrecuente.
Las inmunodeficiencias se clasifican en inmunodeficiencias primarias o congénitas y en inmunodeficiencias secundarias, siendo estas últimas las más frecuentes.
Nos referiremos inicialmente a la relación entre algunas inmunodeficiencias primarias (IDP) y autoinmunidad y finalmente revisaremos infección por VIH y autoinmunidad.
Las IDP son un grupo heterogéneo de más de 150 desórdenes que afectan a distintos componentes del sistema inmune (SI) tanto innato como adaptativo. Aunque las IDP son consideradas como enfermedades raras, se cree que entre el 70 a 90% de éstas son sub diagnosticadas. Se estima que actualmente 1 en 8000 a 1 en 10000 personas tiene una IDP. La prevalencia actual de algunas de las IDP más frecuentes es de 1 en 250 a 1 en 500 (1).
Las IDP usualmente se manifiestan como infecciones frecuentes, recurrentes o persistentes y están muy frecuentemente asociadas con autoinmunidad y/o formación de autoanticuerpos.
La autoinmunidad en las IDP se debería a la alteración de los mecanismos que normalmente regulan negativamente la respuesta inmune.
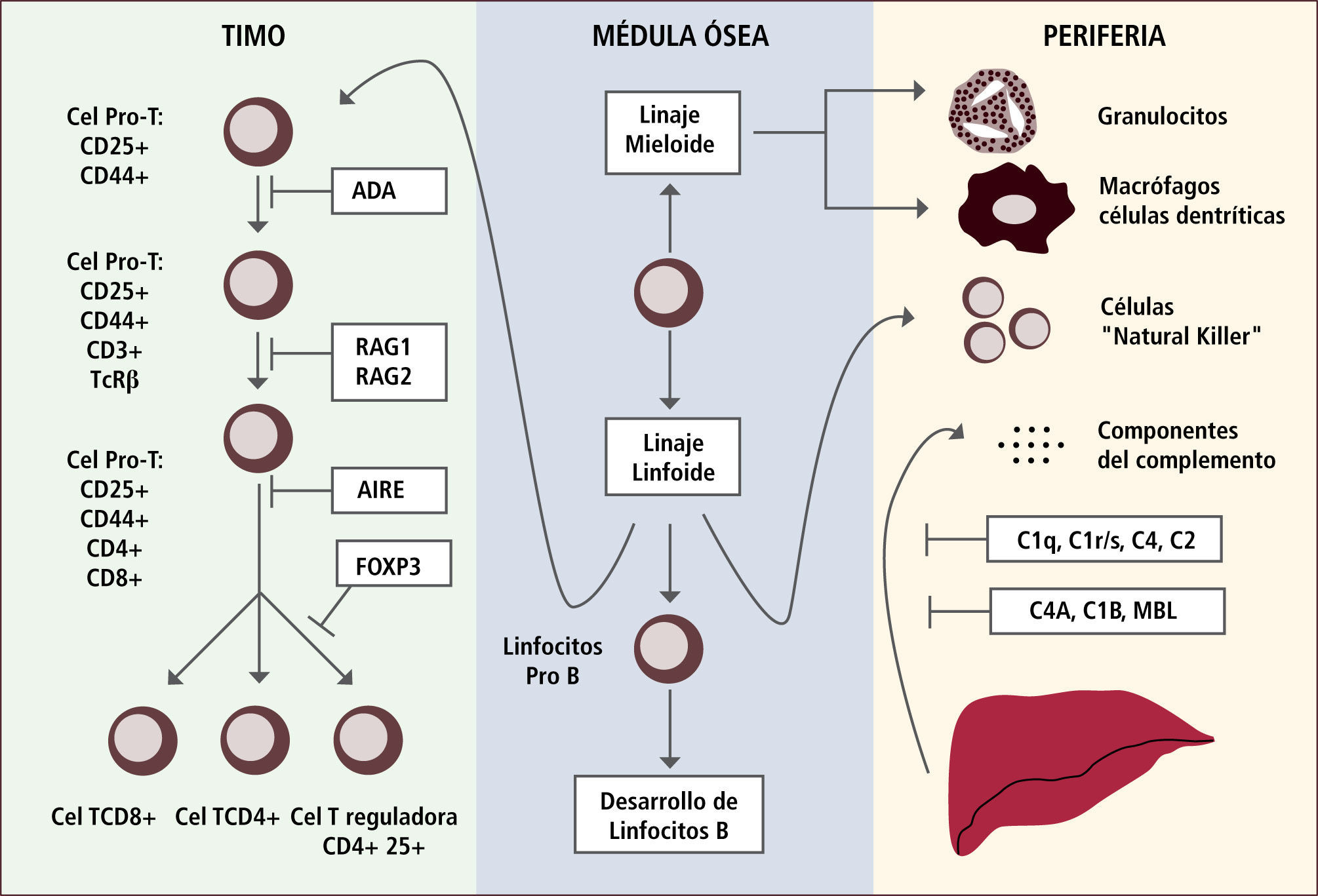
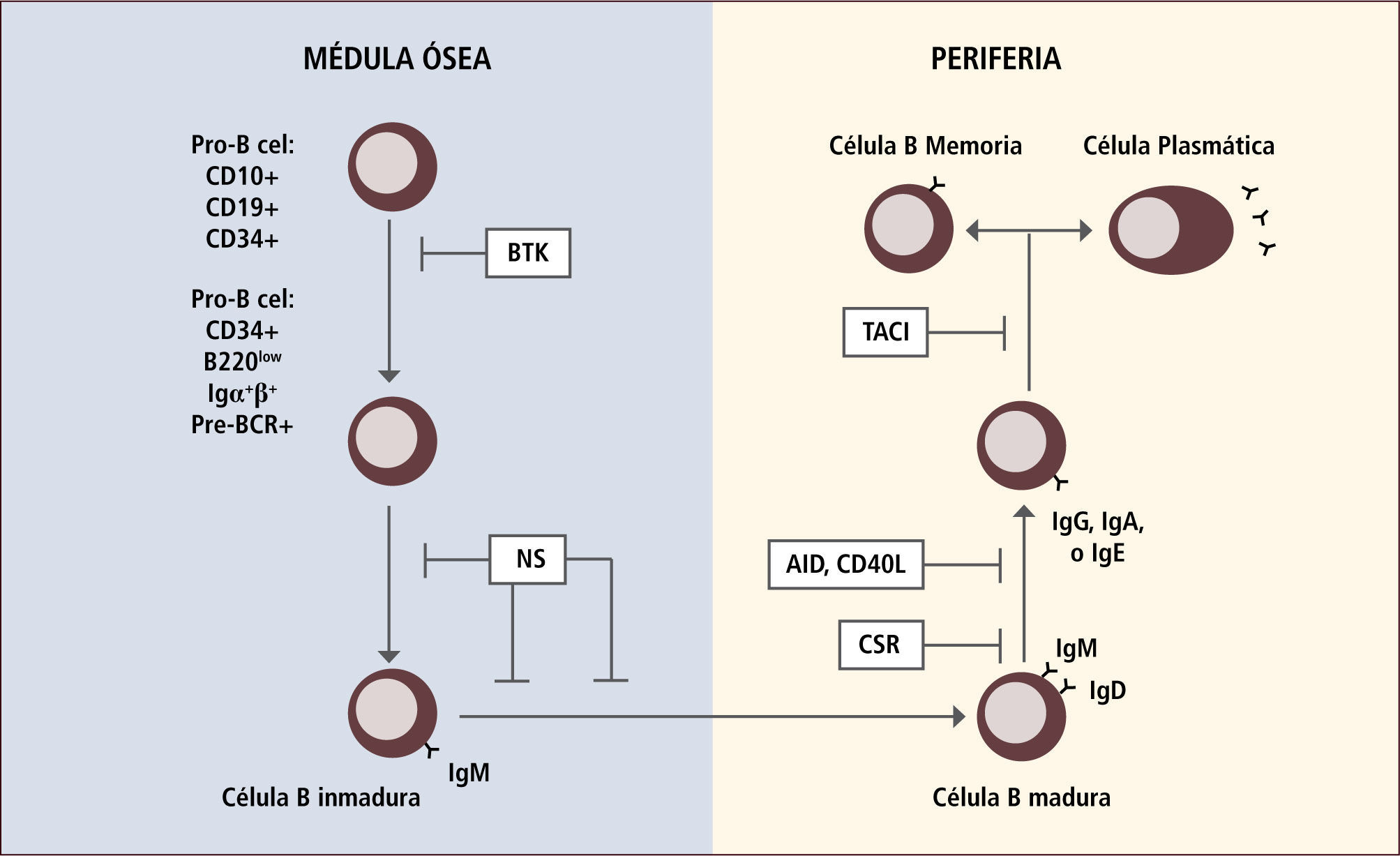
En algunas de las IDP la presencia de autoinmunidad se debe a un defecto en la inducción de tolerancia T periférica, la cual se manifiesta con una disminución de los linfocitos T reguladores (Figura 1). En otras IDP la autoinmunidad se debe a un defecto en la tolerancia de células B ya sea central o periférica (Figura 2).

Ejemplos de defectos génicos en el desarrollo de linfocitos t o del sistema del complemento que llevan al desarrollo de autoinmunidad
Traducido de: Tomado de de Arason Gjet al. Primary Immunodeficiency and Autoimmunity: Lessons from Human Disease. Scandinavian Journal of Immunology 2010, 71:317-328.

Ejemplos de defectos génicos en el desarrollo de linfocitis b que llevan a autoinmunidad
Traducido de: Arason Gjet al. Primary Immunodeficiency and Autoimmunity: Lessons from Human Disease. Scandinavian Journal of Immunology 2010, 71:317-328.
NS: Selección negativa
BTK: Tirosin kinasa de Bruton
CSR: Switch de clases
AID: defecto en activación inducida por deaminasa citidina
CD40L: ligando CD40
TACI: activador de transmembrana
Defectos en selección negativa llevan al desarrollo de ALPS.
Se ha demostrado la existencia de una fuerte correlación entre varias enfermedades autoinmunes y una predisposición familiar (2).
IIDP definidas por la ocurrencia de enfermedades autoinmunes1)Síndrome de Poliendocrinopatía autoinmune - Candidiasis - Distrofia EctodérmicaEste síndrome tiene una herencia de tipo autosómica recesiva y se define por al menos 2 de los siguientes síntomas: candidiasis mucocutánea crónica, hipoparatiroidismo y/o enfermedad de Addison. La primera manifestación es la candidiasis mucocutánea que se presenta alrededor de los 5 años, seguida por el hipoparatiroidismo antes de los 10 años y la falla adrenal antes de los 15 años. Otras de las enfermedades autoinmune órgano específicas que se encuentran en este síndrome incluyen el hipotiroidismo, hipogonadismo, diabetes mellitus (DM) tipo 1, hepatitis autoinmune, anemia perniciosa, vitíligo, alopecía, cirrosis biliar primaria y displasia ectodérmica.
Se debe a un defecto en el gen regulador autoinmune (AIRE), el cual está involucrado en la expresión de antígenos tisulares periféricos del timo.
En condiciones normales este gen incrementa la transcripción de estos antígenos y favorece la selección negativa de linfocitos T autorreactivos, llevándolos a la deleción, impidiendo de esta forma la autoinmunidad.
2)Síndrome linfoproliferativo autoinmune (ALPS)Es un desorden caracterizado por una sobrevida anormal de los linfocitos causada por una disregulación de la vía apopotótica Fas. Sobre el 70% tiene mutaciones en el gen Fas.
Las mutaciones en este gen tienen una penetrancia variable, es así como existen familiares con la misma alteración genética y ausencia o mínima presencia de alteraciones.
Fue descrita en los años 1990 y desde entones se han reportado muchos casos.
Estos pacientes presentan linfoproliferación no maligna crónica, enfermedad autoinmune y neoplasias secundarias (3). La linfoproliferación se manifiesta con linfoadenopatías, esplenomegalia y /o hepatomegalia. La mayoría presenta esta manifestación a temprana edad.
La autoinmunidad es el segundo hallazgo más frecuente, cerca del 70% desarrolla algún tipo de enfermedad autoinmune, dentro de las más frecuentes están las citopenias autoinmunes (anemia hemolítica, trombocitopenia, neutropenia), lo cual se debe a la persistencia de linfocitos B autorreactivos dirigidos contra antígenos encontrados en la médula ósea o en la circulación (2). La severidad es variable y puede ir desde alteraciones de laboratorio hasta compromiso severo de distintos órganos.
Otras manifestaciones menos frecuentes pueden simular un lupus eritematoso sistémico (LES): nefritis, gastritis, hepatitis, urticaria, artritis, colitis y fibrosis pulmonar (3, 4).
Se han descrito manifestaciones neurológicas muy infrecuentes como Síndrome de Guillain Barré, mielitis transversa o ataxia cerebelar autoinmune
3)Síndrome IPExInmunodesregulación, poliendocrinopatía, enteropatía, ligado a X.
Es un síndrome raro, debido a una mutación en el gen Foxp-3, resultando en un desarrollo defectuoso de los linfocitos T reguladores (Treg), por lo que se caracteriza por activación de linfocitos T y aumento en la producción de citoquinas.
Clínicamente se manifiesta por enteritis autoinmune, DM tipo 1 durante los primeros meses de vida, eccema, hipotiroidismo, anemia hemolítica autoinmune (AHAI), nefropatía membranosa e infecciones recurrentes.
IIIDP asociadas a manifestaciones autoinmunes1)Linfopenia CD4 idiopática (LCI)Se caracteriza por un recuento de linfocitos T CD4 < a 300 células/mm3, en ausencia de infección por VIH u otra agente viral, otras causas de inmunodeficiencias y/o de drogas (5). Estos pacientes tienen mayor susceptibilidad a presentar infecciones por Criptococcus neoformans, virus papiloma y micobacterias atípicas.
Algunos de estos pacientes presentan manifestaciones autoinmunes ya sea antes del diagnóstico de la LCI o durante el seguimiento de ésta, por ejemplo: LES, síndrome antifosfolípidos, enfermedad de Basedow Graves, AHAI, colitis ulcerosa y vitíligo (6).
En el síndrome de Sjögren algunos pacientes presentan linfopenia CD4 idiopática, la que se asocia a la presencia de anticuerpos anti CD4. Tienen un bajo recuento de linfocitos T CD4 y un alto recuento de linfocitos T CD8, esta inversión del índice se asocia a enfermedad renal severa y trombocitopenia (5).
2)Inmunodeficiencia Común Variable (IDCV)Es la inmunodeficiencia sintomática más frecuente, con una incidencia de 1 en 25.000 a 1 en 66. 000. Se han descrito diversos defectos genéticos asociados a esta enfermedad.
Generalmente se diagnostica en la segunda y cuarta década de la vida, siendo frecuente el retraso entre la aparición de los primeros síntomas y el diagnóstico de certeza.
Se caracteriza por la presencia de niveles reducidos de Inmunoglobulina G (IgG), Inmunoglobulina A (IgA) y/o Inmunoglobulina M (IgM) comparado con los estándares para la edad, acompañado por una alteración o ausencia en la producción de anticuerpos. Alrededor del 70 a 80% de los pacientes tienen infecciones sinopulmonares recurrentes, otros presentan infecciones gastrointestinales, malabsorción, linfoadenopatías, esplenomegalia, neoplasias y enfermedades autoinmunes (7).
Las enfermedades autoinmunes afectan aproximadamente al 20% de las IDCV y son comúnmente la primera manifestación de IDP (5-7), con un leve predominio en el sexo femenino, la mayoría son órgano específicas (6, 7) (Tabla 1).
Manifestaciones autoinmunes en IDCV
| Hematológicas: |
|---|
| • Citopenias autoinmunes |
| • Anemia hemolítica autoinmune |
| • Púrpura trombocitopénica idiopática |
| • Neutropenia autoinmune |
| Gastrointestinales: |
|---|
| • Anemia perniciosa |
| • Gastritis atrófica |
| • Enfermedad celíaca |
| • Cirrosis biliar primaria |
| • Enfermedad inflamatoria intestinal |
| Reumatológicas: |
|---|
| • Síndrome de Sjögren |
| • Lupus eritematoso sistémico |
| • Artritis Reumatoide |
| • Artritis Crónica Juvenil |
| • Vasculitis |
| Endocrinológicas: |
|---|
| • Tiroiditis de Hashimoto |
| • Diabetes Mellitus tipo 1 |
| Dermatológicas: |
|---|
| • Vitiligo |
| • Alopecía |
| Neurológicas: |
|---|
| • Síndrome de Guillain Barré |
Manifestaciones autoinmunes hematológicas: son las más frecuentes: Púpura trombocitopénico inmune (PTI) y AHAI ocasionalmente se presentan juntos en la forma de síndrome de Evans. La prevalencia de uno u otra de estas manifestaciones en IDCV se estima en 5 a 8% (6-8). La linfopenia y/o neutropenia autoinmunes son muy infrecuentes.
Manifestaciones gastrointestinales: Enfermedad inflamatoria intestinal, gastritis atrófica, cirrosis biliar primaria y enfermedad celíaca.
Manifestaciones reumatológicas: Las manifestaciones articulares que simulan una artritis reumatoide (AR) o una artritis reumatoide juvenil ocurren en el 1 a 10% de los pacientes con IDCV (7). Se caracteriza por una oligo o poliartritis simétrica de rodillas, tobillos y manos y puede resultar en destrucción articular, las alteraciones histológicas de la membrana sinovial son distintas a las encontradas en pacientes con AR: hay hiperplasia sinovial, proliferación de capilares sin un infiltrado importante de linfocitos o polimorfonucleares, sin linfocitos B y el infiltrado de linfocitos T está compuesto por LT CD8+.
Hay ausencia de anticuerpos antinucleares (ANA) o de Factor Reumatoide (FR) debido a la carencia de anticuerpos (7).
El compromiso monoarticular en la IDCV habitualmente es secundario a infección por Streptococcus pneumoniae, Haemophylus influenzae o Staphylococcus aureus o menos frecuentemente por gérmenes atípicos.
El LES ha sido descrito en un 1% o menos de las IDCV. La mayoría ha desarrollado el LES antes de las manifestaciones clínicas de la inmunodeficiencia, de hecho al momento del diagnóstico del LES tenían recuento de inmunoglobulinas normales y posteriormente desarrollaron la hipogammaglobulinemia. Algunos de estos pacientes persisten con ANA y /o anticuerpos anti DNA y anticardiolipinas positivas (8, 9).
Otras enfermedades autoinmunes asociadas a IDVC: Anemia perniciosa, tiroiditis de Hashimoto, alopecía y vitíligo.
En todos los pacientes con IDCV debe realizarse un estudio de LB de memoria, ya que se ha demostrado que aquellos que tienen menos de un 0.4% de LB de memoria de switch pero un número importante de linfocitos B inmaduros tienen mayor predisposición a tener enfermedades autoinmunes y esplenomegalia, estos pacientes no deben ser esplenectomizados ya que tienen mayor riesgo de desarrollar infecciones por gérmenes capsulados (10, 11).
Siempre debe realizarse un recuento de inmunoglobulinas como parte del estudio de un paciente con alguna enfermedad autoinmune sobre todo en las citopenias autoinmunes para descartar una IDCV (12).
3)Déficiencia selectiva de Inmunoglobulina AEs la IDP más común con una frecuencia de 1 en 400 a 1 en 700 en caucásicos. Se define como un desorden caracterizado por recuento de IgA sérica < a 7mg/dl con IgM e IgG dentro de rango normal en individuos sobre los 4 años de edad (13).
La mayoría de estos pacientes son clínicamente asintomáticos, sin embargo aquellos que presentan síntomas se manifiestan con infecciones respiratorias y gastrointestinales recurrentes, alergias y autoinmunidad. Aquellos pacientes que tienen asociado un déficit de subclases de IgG son más susceptibles a presentar infecciones (13, 14).
Las manifestaciones autoinmunes más frecuentes son las siguientes: Enfermedad de Graves, se ha descrito una mayor incidencia de esta enfermedad en los pacientes con déficit de IgA.
LES se ha reportado con una frecuencia aumentada en los pacientes con Déficit de IgA. A la inversa también se ha reportado una frecuencia de déficit de IgA aumentadas en pacientes con LES de 1 en 19 en Estados Unidos y de 1 en 130 en España (14).
DM tipo 1: Se ha reportado una prevalencia de Défcit de IgA en pacientes DM tipo1 de 1 en 27 a 1 en 261 (14), indicando una frecuencia mayor a la observada en población general.
Enfermedad celíaca: se ha reportado una prevalencia de Déficit de IgA en enfermedad celíaca de 1 en 39, y también una mayor prevalencia de enfermedad celíaca en pacientes con Déficit de IgA (14-16).
Miastenia Gravis (MG): la prevalencia de Déficit de IgA en pacientes con MG es de 1 en 221 pacientes (14).
AR: la prevalencia de Déficit de IgA en AR es de 1 en 194, en Artritis Reumatoide juvenil es de 1 en 37.
Se piensa que en el Déficit de IgA la cual está presente desde el nacimiento, el aumento de la frecuencia de infecciones podría desencadenar algunas enfermedades autoinmunes como la enfermedad celíaca y el LES. Sin embargo, el inicio del Déficit de IgA en algunos pacientes con enfermedad celíaca se ha reportado posterior al inicio de los síntomas gastrointestinales, en estos casos el background genético asociado al Complejo mayor de histocompatibilidad (MHC) sería el principal factor contribuyente (14). Según estudios recientes se plantea que el Déficit de IgA sería también una enfermedad autoinmune (14, 17).
4)Síndrome de Hiper IgMEste síndrome se caracteriza por un defecto en la vía del ligando CD40 o de otras proteínas requeridas para el switch de clases de las inmunoglobulinas. Estos pacientes tienen ausencia o niveles séricos bajos de IgG e IgA con IgM elevada o normal y una mayor predisposición a presentar infecciones recurrentes sinopulmonares y gastrointestinales e infecciones por gérmenes oportunistas.
Se describen diversas enfermedades autoinmunes como: enfermedad inflamatoria intestinal en el 6% de los pacientes, artritis seronegativas en el 11%, neutropenia en el 45% al 60%, anemia en el 15% y trombocitopenia en el 4%, así como también hepatitis autoinmune y colangitis.
5)Síndrome de GoodEsta es una rara asociación entre timoma e inmunodeficiencia que se presenta entre los 40 y los 70 años de edad, se caracteriza por la presencia de timoma benigno, linfocitos B bajos o ausentes en sangre periférica, hipogammaglobulinemia, linfopenia CD4 e inversión del índice CD4/CD8 (18, 19). Se manifiesta con infecciones recurrentes principalmente por patógenos intracelulares. El 50% de los pacientes presenta alguna enfermedad autoinmune como la Miastenia Gravis y /o algunas citopenias como neutropenia y aplasia de glóbulos rojos, con menor frecuencia se ha descrito LES, DM, colitis ulcerosa, colangits esclerosante, pénfigo, liquen plano, síndrome de Sjögren y polimiositis (6, 18, 19).
Hasta el 30% los pacientes presenta ANA positivos (18).
En todo paciente con timoma debe descartarse la presencia de inmunodeficiencia y/o autoinmunidad.
6)Síndrome de Wiskott Aldrich (WAS)Este es un síndrome ligado a X que se inicia en la infancia y se caracteriza por la presencia de eccema, infecciones recurrentes y trombocitopenia. Estos pacientes son deficientes en interleuquina 2 (IL-2), citoquina requerida para la sobrevida de los linfocitos Treg (5).
El 40 al 70% de los pacientes puede presentar manifestaciones autoinmunes, la mayoría de ellos más de una, asociada a altos títulos de autoanticuerpos (2, 6).
La manifestación más frecuente es la AHAI, también se describe la neutropenia autoinmune, artritis, vasculitis, uveítis, glomerulonefritis y enfermedad infamatoria intestinal (5, 6).
7)Déficit hereditario de proteínas del sistema del complementoLas deficiencias de los componentes tempranos del complemento frecuentemente se asocian al desarrollo de enfermedades autoinmunes mediadas por complejos inmunes. Esto se observa en el 93% de los pacientes con deficiencia de C1q, el 60% de los pacientes con deficiencia de C1s o C1r, el 90% de los pacientes con deficiencia de C4 y el 10% de los pacientes con deficiencia de C2 y de C3 (2, 19). Esto refleja la importancia de los componentes tempranos de la vía clásica en la eliminación segura de los complejos inmunes y de células apoptóticas. La acumulación de estas últimas deriva en un aumento de liberación de antígenos nucleares y gatilla la producción de anticuerpos antinucleares. También se postula que los anticuerpos antinucleares serían secundarios al daño tisular como resultado del depósito de complejos inmunes en los tejidos (2).
La deficiencia de C1r, C1s y C1q son raras y se asocian a un mayor riesgo de desarrollar LES pediátrico. Más del 90% de los pacientes homozigotos para deficiencia de C1q presentan LES like, mientras que el 50% de los pacientes deficientes de C1r y C1s presentan LES (6).
La deficiencia completa de C4 se asocia a LES, Púrpura de Schönlein Henoch, síndrome de Sjögren y glomerulonefritis. La deficiencia de C3 se asocia a LES y vasculitis (2, 6).
La ausencia de C1 inhibidor se asocia a angioedema hereditario y en algunos casos a nefritis lúpica y otras manifestaciones autoinmunes. Las deficiencias de los componentes tardíos del complemento se asocian más bien a infecciones y no a autoinmunidad.
La deficiencia de lectina de unión a la manosa (MBL) es la inmunodeficiencia más común con un 5% de la población afectada, su fenotipo es habitualmente leve y se asocia a una mayor prevalencia de LES e infecciones (19).
8)Enfermedad granulomatosa crónicaEsta enfermedad se caracteriza por una producción disminuida o ausente de NADPH oxidasa por parte de los neutrófilos y se manifiesta con infecciones bacterianas y por hongos recurrentes con formación de granulomas en pulmón, piel o tracto gastrointestinal. Se asocia a una mayor predisposición a presentar lupus discoide incluso en los portadores asintomáticos (5, 19).
IIIManifestaciones autoinmunes asociadas a infección por virus de la inmunodeficiencia humana (VIH)Manifestaciones reumatológicas: Son las manifestaciones de autoinmunidad más frecuentes en estos pacientes (20).
Autoanticuerpos: se describen una serie de autoanticuerpos habitualmente a títulos bajos como por ejemplo: FR, ANA, anticuerpos anticardiolipinas, anticoagulante lúpico, anticuerpos anti citoplasma de neutrófilos (ANCA), anticuerpos anti DNA, anti Sm, anti U1RNP, anti La. Se piensa que se producen por reactividad cruzada entre los antígenos virales y autoantígenos, aparecen en pacientes con enfermedad avanzada por lo que su presencia es de mal pronóstico (20).
Espondiloartropatías (EAA)Es la manifestación reumatológica más común en la infección por VIH, de hecho su prevalencia ha aumentado en los países con una alta incidencia de esta infección (20-22).
Las características clínicas de la artritis reactiva y el Síndrome de Reiter son similares en los pacientes no infectados por el VIH pero el dolor lumbar, la sacroileitis y la uveítis son infrecuentes. Se puede manifestar en cualquier etapa de la enfermedad. El HLA- B27 es positivo en el 70% de los pacientes caucásicos (20).
Psoriasis y artropatía psoriática: Ocurre habitualmente en etapas tardías de la enfermedad, pero también puede preceder el inicio de la inmunodeficiencia hasta en el 10% de los casos. Su presentación es más bien poliarticular y asimétrica e infrecuentemente se comprometen las articulaciones sacroilíacas.
La infección por HIV puede exacerbar la psoriasis, de hecho su presencia se asocia a peor pronóstico (20).
Espondiloartropatía indiferenciada: Es la forma más común de EAA asociada a VIH, tiene algunas características de artritis reactiva o de artropatía psoriática.
El tratamiento de la EAA es complejo debido a que el uso de metotrexato u otros inmunosupresores pueden acelerar la evolución del VIH hacia etapa SIDA.
Artritis asociadas a VIH: puede presentarse en cualquier etapa de la infección, habitualmente es mono u oligoarticular y compromete extremidades inferiores. El líquido sinovial es no inflamatorio. El HLA-B27 y los autoanticuerpos son negativos. Habitualmente es autolimitada.
Artralgias asociadas a VIH: se describen en el 2 al 45% de los pacientes en cualquier etapa de la enfermedad y son oligo o poliarticulares.
Polimiositis asociada a VIH: es clínicamente indistinguible de la polimiositis idiopática. Incluso se han descrito dermatomiositis.
Síndrome de linfocitosis infiltrativa difusa o síndrome de Sjögren- like se caracteriza por aumento de volumen de glándulas salivales, linfocitosis CD8 periférica, síndrome sicca y manifestaciones extraglandulares con autoanticuerpos negativos (FR, anti Ro y anti La).
La Zidovudina reduce eficazmente el aumento de volumen glandular y los síntomas del síndrome sicca (20).
Síndrome de Lupus like asociado a VIH: comparte muchas características con el LES. A su vez también se pueden encontrar anticuerpos anti VIH falsos positivos en pacientes con LES. Es importante poder diferenciar y aclarar bien el diagnóstico para implementar un tratamiento adecuado y oportuno.
Vasculitis asociada a VIH: son infrecuentes, se describen en menos de un 1% de los pacientes (23). Dentro de éstas la más frecuente es la Poliarteritis nodosa (PAN) - like, se presenta con manifestaciones distintas a la PAN (20, 23).
Síndrome Kawasaki- like: A diferencia de la enfermedad de Kawasaki, esta se presenta sin compromiso coronario y se asocia a compromiso gastrointestinal y cefalea, responde bien al tratamiento con gammaglobulina (24).
Enfermedad renal por Complejos inmunes (CI) en VIHLa glomerulonefrtis por CI es común en las biopsias renales de pacientes con infección VIH, se describen dentro de este grupo: la nefropatía por IgA, glomerulonefritis membranoproliferativa, nefropatía membranosa, glomerulonefritis de tipo lúpica y glomerulopatía inmunotactoide, esto se debería a la hipergammaglobulinemia policlonal de la infección VIH y la formación de CI circulantes secundario a la respuesta inmune frente a los gérmenes oportunistas (25).
ConclusionesLas manifestaciones autoinmunes son frecuentes y múltiples en pacientes con inmunodeficiencias. El diagnóstico de ID debe ser hecho lo más precozmente posible para evitar las complicaciones y secuelas.
Es importante considerar a las inmunodeficiencias dentro del diagnóstico diferencial de las enfermedades autoinmunes, especialmente si hay historia de infecciones recurrentes.
Debido a que las enfermedades autoinmunes pueden ser la primera manifestación de una IDP, debe realizarse siempre dentro del estudio un recuento de inmunoglobulinas inicial y durante el seguimiento para pesquisar también aquellas hipogammaglobulinemias que se desarrollan tardíamente en la evolución de la enfermedad.
Dado que las inmunodeficiencias secundarias son las inmunodeficiencias más frecuentes, y dentro de estas la infección por VIH, se debe estudiar siempre ante la sospecha de alguna enfermedad autoinmune sobre todo si su manifestación es atípica o se asocia a infecciones recurrentes.
La autora declara no tener conflictos de interés, con relación a este artículo.