



Los Errores Innatos del Metabolismo (EIM) son un grupo de condiciones caracterizadas por el acúmulo de sustancias tóxicas producido habitualmente por un defecto enzimático. Su relevancia dentro del grupo de las Enfermedades Raras, es que son consideradas hoy un conjunto de patologías tratables, ya que han sido particularmente beneficiadas por leyes de drogas huérfanas, permitiendo el acceso a terapias seguras y eficaces en tratar sus síntomas y la causa que las produce.
Su diagnóstico se debe sospechar clínicamente ante ciertos patrones de síntomas y signos, y con un laboratorio sencillo disponible en todos los hospitales y clínicas de Chile, se puede acceder fácilmente a una aproximación inicial, confirmada por estudios realizados en centros de referencia.
Su tratamiento debe ser instaurado oportunamente para evitar el desarrollo de secuelas irreparables. La existencia de Programas de Pesquisa Neonatal es la aproximación diagnóstica ideal para llegar precozmente al diagnóstico de estas condiciones.
Inborn Errors of Metabolism are a group of conditions characterized by the accumulation of toxic substances commonly produced by an enzymatic defect. Its relevance within the group of rare diseases is that they are considered today a set of treatable pathologies, as they have been particularly benefit from orphan drug laws, allowing access to safe and effective therapies to treat their symptoms.
Its diagnosis should be suspected clinically to certain patterns of symptoms and signs, and with a simple laboratory available in all Hospitals and Clinics Chile can easily earn an initial approximation, to be confirmed by studies in reference centers. The treatment should be started promptly to prevent the development of irreversible sequelae. The existence of Newborn Screening Programs is ideal for reaching early diagnosis and treatment of these conditions.
Los Errores Innatos del Metabolismo (EIM) se manifiestan en la edad pediátrica, desde las primeras horas de vida y hasta la adolescencia o edad adulta con síntomas y signos similares a otras patologías. No reconocerlos conduce a secuelas como desnutrición, convulsiones, retardo mental e incluso la muerte. La prevención de estas secuelas con un diagnóstico oportuno es el desafío al que se enfrentan los pediatras.
La relevancia de este grupo de condiciones en el contexto de las enfermedades raras, se ha marcado en el transcurso de las últimas décadas con el desarrollo de terapias específicas, con efectividad y seguridad probadas por estudios clínicos, que han posicionado a estas patologías como “enfermedades raras tratables”. Desde la aparición de las leyes de drogas huérfanas en la década de los años ‘80 y la tecnología que permitió resolver paradigmas como la producción por ingeniería de genética recombinante, el reemplazo para enzimas deficientes o el diseño de inhibidores de la formación de sustratos tóxicos, este grupo de condiciones ha visto aumentar su visibilidad en la comunidad científica y ha cambiado la forma en que las enfrentamos.
Esta realidad ha llevado a estas enfermedades a transformarse en una punta de lanza que arrastra a otras enfermedades raras a legislaciones que regulan la investigación, el diagnóstico, el registro y acceso a estas drogas denominadas huérfanas.
El permanente avance de los conocimientos en el área de la terapia génica hacen pensar que seguiremos siendo testigos de un número creciente de terapias para nuestros enfermos.
DEFINICIÓNLos EIM son enfermedades monogénicas, de herencia autosómica recesiva en su mayoría. La alteración en un gen produce un defecto enzimático, que conduce a las alteraciones bioquímicas características y a un fenotipo desadaptativo. Hoy se reconocen condiciones dentro del grupo de los EIM donde puede fallar un transportador o modificadores post transcripcionales de la expresión génica, dejando atrás la creencia de que la falla de un gen produce el defecto de una enzima y esta a su vez un solo fenotipo clínico. La pesquisa neonatal ampliada, a su vez, ha permitido conocer amplios espectros de presentación clínica que hace pocos años no sospechábamos y de los que hoy somos testigos. Esto a través de la historia natural para pacientes, que diagnosticados bioquímica o genéticamente en el período neonatal desarrollan síntomas diferentes a los tradicionalmente descritos en formas “clásicas” de presentación.
ASPECTOS CLÍNICOS Y EXAMEN FÍSICOSe distinguen clásicamente cuatro formas de presentación clínica:
1.-Síntomas agudos en el período neonatal: Habitualmente síntomas inespecíficos iniciales como rechazo de la alimentación, vómitos explosivos, apneas (centrales) o episodios que aparentemente ponen en peligro la vida (ALTE), compromiso de conciencia (desde el letargo y somnolencia pudiendo llegar al coma profundo), convulsiones, compromiso hemodinámico y muerte. Se pueden presentar también con edema cerebral y hemorragia intracraneana.
2.-Síntomas agudos y recurrentes de inicio más tardío: Pueden presentarse después del período de recién nacido en relación a cambios nutricionales o la presencia de infecciones, o bien, aparecer en adolescentes o adultos jóvenes en relación a gatillantes como ingesta excesiva de proteínas, menarquia, cirugías o cualquier otro evento que produzca un stress metabólico importante. Cada episodio puede derivar en mejoría espontánea o en muerte inexplicada. Durante el período entre las crisis el paciente parece normal clínica y bioquímicamente.
Frente al niño que presenta compromiso de conciencia se debe descartar un EIM, así como ante los diagnósticos de encefalitis, jaqueca o intoxicación, especialmente cuando el paciente presenta cetoacidosis, hiperamonemia o acidosis láctica. Estos síntomas se pueden acompañar de un amplio espectro de signos hepáticos, digestivos, neurológicos o psiquiátricos.
3.- Síntomas crónicos y progresivos: Muchas veces inadvertidos por años, podemos encontrar síntomas gastrointestinales (anorexia, vómitos y retardo del crecimiento) y neurológicos (retardo del desarrollo psicomotor, convulsiones, trastornos motores y cognitivos), como los de presentación más habitual.
4.- Síntomas específicos y permanentes característicos de los EIM: Hallazgos tales como miocardiopatía dilatada o hipertrófica, hepatoesplenomegalia, alteraciones del cristalino o dismorfias pueden orientar a EIM específicos.
Se debe hacer notar que gracias al avance en el conocimiento de los espectros de manifestación clínica, hoy sabemos que estas condiciones se pueden presentar de forma prenatal, como es el caso de la relación entre el Síndrome de HELLP –un cuadro obstétrico que afecta a la madre caracterizado por hemolisis, enzimas hepáticas elevadas, plaquetas bajasy la presencia de un defecto de beta oxidación de ácidos grasos en el niño en gestación, o la presencia de convulsiones intra útero en el déficit de piridoxina, relatadas por las madres cuando observan las convulsiones que aparecen precozmente en la primeras horas de vida. Así también, pacientes con el mismo defecto enzimático, incluso la misma mutación, pueden presentar formas clásicas de una enfermedad o no manifestar ningún síntoma.
DIAGNÓSTICO DIFERENCIALEn el recién nacido con presentación aguda, la presencia de infección no descarta un EIM ya que ésta puede ser el gatillante, o bien el mismo EIM puede aumentar la predisposición a tener infecciones.
El carácter multisistémico y progresivo de los EIM hacen necesario reconocer patrones de síntomas, variables de paciente a paciente y cambiantes en el tiempo, más que enfermedades relacionadas a un único síntoma.
El aspecto más relevante, por tanto, es un alto nivel de sospecha clínica; esto es, no considerar a este grupo de condiciones como un diagnóstico de descarte, sino plantear su presencia al comienzo del estudio de síntomas y signos frecuentes, en particular cuando se presentan en un patrón reconocible.
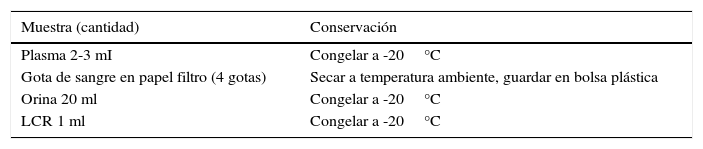
LABORATORIO E IMÁGENESEl laboratorio inicial debe ir orientado a encontrar metabolitos acumulados (amonio, ácido láctico, aminoaciduria, aminoacidemia), o a consecuencias de cada EIM según sea el caso (hemograma con recuento de plaquetas, orina completa, gases venosos, electrólitos plasmáticos, glicemia, cetonemia/cetonuria). Considerando el carácter intermitente de algunos EIM, se debe considerar la necesidad de tomar una “muestra crítica” en el Servicio de Urgencia. Esta muestra permitirá solicitar exámenes específicos una vez recibidos los resultados de los exámenes iniciales y luego de haber observado la evolución clínica.
MUESTRA CRÍTICACon estas muestras se pueden solicitar exámenes tales como Espectrometría de Masa en Tandem (perfil de aminoácidos y acilcarnitinas), niveles de carnitina, ácido pirúvico, cuantificación de aminoácidos, ácidos orgánicos en orina (realizados en Chile por el Laboratorio de Enfermedades Metabólicas del INTA, U de Chile), o algunos otros como las acilglicinas, ácidos grasos de cadena muy larga (enviados a centros de referencia fuera del país).
TRATAMIENTOA. Tratamiento de los EIM en el recién nacido y lactante
1.- Inmediato:
- •
Practicar exámenes generales y tomar “muestra crítica”.
- •
Soporte vital y estabilización clínica.
- •
Mantención hidroelectrolítica y del equilibrio ácido-base: corregir la acidosis metabólica con bicarbonato si el pH es 7,10 o bicarbonato 10 mEq/l.
2.-Evitar producción endógena de metabolitos tóxicos y favorecer anabolismo:
Régimen cero en las primeras 24 horas, suero glucosado 10% (según hipoglicemia o por ingesta calórica aportando 6-8mg/k/min. de carga de glucosa inicialmente). Considerar que el ayuno prolongado es perjudicial en la mayoría de los EIM y debe ser mantenido el menor tiempo posible.
Tan precoz como el segundo día de vida, aportar triglicéridos de cadena mediana para prevenir catabolismo protéico (0,5-2g/k/día). Se debe proceder de esta forma si el amonio sobrepasa los 300 ug% y si hay una acidosis metabólica con grave cetoacidosis. Tanto los carbohidratos como los lípidos se deben dar inicialmente por vía endovenosa, pero se debe usar la vía enteral lo más pronto posible (mezclas de polímeros de glucosa y triglicéridos de cadena mediana utilizando una bomba de infusión continua). Iniciar fórmulas metabólicas específicas según diagnóstico confirmado.
3.-Suplemento de sustratos:
- •
L-Carnitina a todos los pacientes, en dosis de 150-300mg/kg/día por vía endovenosa u oral, ya sea en infusión continua o fraccionada en 3 dosis. No se conocen condiciones donde el uso de carnitina pueda ser perjudicial, por lo que su uso en el manejo de urgencia esta ampliamente aceptado.
- •
Clorhidrato de arginina al 10% también se prescribe en todos los casos de hiperamonemia hasta no aclarar etiología (dosis: 0,6g/k a pasar en 90 minutos endovenoso). Su uso en argininemia no estaría indicado.
- •
Las vitaminas que se suplementan son Biotina (10mg/día oral o por SNG), Tiamina (50mg/día), Riboflavina (100mg/ día).
- •
Como un ejemplo, en la acidemia metilmalónica se indica vitamina B12 (1-2mg IM). Terapias más específicas de la enfermedad se inician cuando se tenga diagnóstico definitivo y consisten básicamente en restringir los compuestos específicos involucrados en el defecto enzimático. Si los exámenes fueran negativos, a las 48 horas aportar leche materna o fórmula de bajo contenido proteico.
4. Remoción de sustancias tóxicas: El tratamiento nutricional intensivo y la suplementación de sustancias descritas debieran en muchos casos mejorar el cuadro. En caso de que esto no ocurra en 24-48 horas, se debe considerar la remoción de sustancias tóxicas con diálisis.
5.- Indicaciones de diálisis:
- •
Amonemia 500 ug/dl (hiperamonemia severa)
- •
Compromiso de conciencia progresivo.
- •
Convulsiones
- •
Coma
La mejoría clínica con diálisis aparece en forma variable, a las 24–48 horas en las acidurias orgánicas y entre 3-7 días en las alteraciones del ciclo de la urea o enfermedad orina olor a jarabe de arce.
B. Otras terapias: En los últimos años han aparecido nuevas alternativas terapéuticas que han transformado a este grupo de patologías en tratables en su mayoría, lo que exige al pediatra mantenerse informado del rápido avance en esta área. Además de los enfoques ya mencionados, hoy contamos con Terapia de Reemplazo Enzimático (Enf. de Gaucher, Fabry, Pompe, Mucopolisacaridosis I, II, IVA y VI), Terapia de Inhibición de Sustrato (Tirosinemia tipo I) y una activa línea de producción en diferentes compañías de biotecnología que ofrece a nuestros pacientes la alternativa de estudios clínicos cuando no contamos con terapias específicas. El trasplante de médula ósea aún es una alternativa para algunas enfermedades como la adrenoleucodistrofia ligada al X o en pacientes con mucopoliscaridosis I de menos de dos años de vida.
DERIVACIÓN A ESPECIALISTASe debe solicitar evaluación por especialista en Enfermedades Metabólicas a todo niño en quien se considere un EIM.
La sospecha clínica precoz por parte del pediatra es fundamental, así como la instauración de Programas de Pesquisa Neonatal Ampliada, que permitan extender los beneficios del programa hoy existente en Chile para fenilcetonuria.
Hoy en día, solo algunos Centros Privados de Salud ofrecen el envío de exámenes a laboratorios de referencia para la realización de una pesquisa ampliada. La proyección es que en el futuro cercano se pueda contar con un Programa Nacional.
Los autores declaran no tener confl de interés, en relación a este artículo.