




Las displasias esqueléticas son un grupo heterogéneo de enfermedades caracterizadas por la alteración primaria del tejido óseo y/o cartilaginoso.
La incidencia de muchas de estas entidades es desconocida, estimándose una incidencia general de 1 de 4.000 recién nacidos vivos.
Frente a pacientes con restricción prenatal de crecimiento o que presentan talla baja durante la niñez, especialmente si existe desproporción corporal, debemos sospechar la presencia de una displasia esquelética. El estudio radiológico es fundamental para confirmar la afección ósea e intentar aproximarse a un diagnóstico preciso, pero requiere de especialistas expertos. En la actualidad contamos con estudio molecular confirmatorio y son las alteraciones de los genes FGFR3, COL2α1 y SHOX los que dan cuenta de las displasias que más frecuentemente observaremos en nuestra práctica clínica.
En los últimos años la mayor precisión diagnóstica se ha acompañado de mayores oportunidades terapéuticas. El desarrollo de nuevas técnicas quirúrgicas de apoyo en casos de deformidades óseas y de técnicas menos invasivas de alargamiento han determinado mejoría en talla final, pero por sobre todo, en la calidad de vida de nuestros pacientes.
The skeletal dysplasias are a heterogeneous group of diseases characterized primarily by impaired primary bone and/or cartilage development.
The incidence of many of these entities is unknown; an overall incidence of 1 in 4,000 live births is estimated.
Clinically, we must suspect the presence of a skeletal dysplasia in patients with prenatal growth restriction or childhood with short stature, especially in the presence of body disproportion. The radiological study is essential to confirm the bone condition so to try approach an accurate diagnosis, and skilled experts are required. Today we have the possibility to confirm the diagnosis by molecular studies, and we know that molecular alterations in FGFR3, COL2α1 and SHOX genes account for the most frequent cases that we will observe in our clinical practice.
In recent years the increased diagnostic accuracy has been accompanied by major therapeutic opportunities. The development of new surgical techniques in bone deformity management and less invasive enlargement techniques have certainly improved the final height, but above all, in the quality of life of our patients.
Las displasias esqueléticas (DE) son un grupo heterogéneo de enfermedades que afectan primariamente al tejido óseo y/o cartilaginoso.
Son heterogéneas en su forma de presentación clínica, el patrón de herencia, el compromiso radiológico y en la base molecular subyacente.
La incidencia de muchas de estas patologías es desconocida, estimándose una incidencia general de 1 de 3.000 - 4.000 recién nacidos vivos. Es probable, por lo tanto, que todo pediatra se vea enfrentado al manejo de pacientes con alguna Displasia Esquelética en su práctica clínica1.
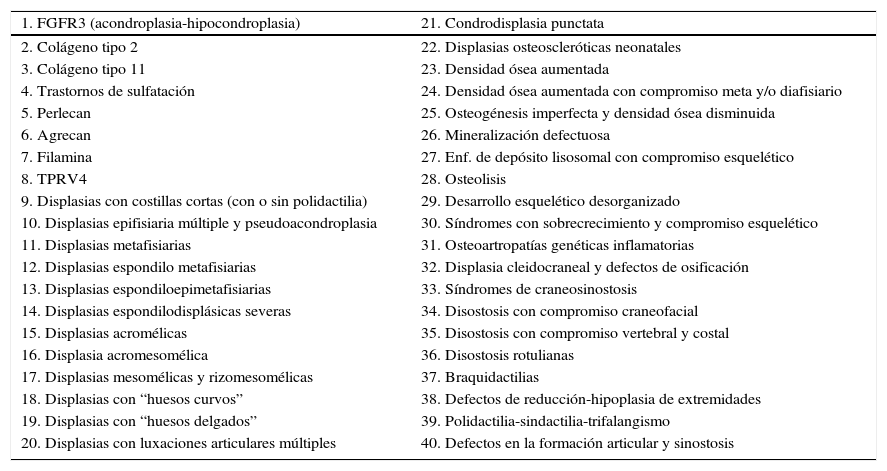
La Sociedad Internacional de Displasias Esqueléticas elaboró en el año 2010 la última Clasificación de Displasias Esqueléticas, en la que se identificaron 456 entidades agrupadas en 40 categorías en función de sus características bioquímicas, radiológicas y moleculares (Tabla 1)2.
Clasificación de las displasias esqueléticas en 40 categorías
| 1. FGFR3 (acondroplasia-hipocondroplasia) | 21. Condrodisplasia punctata |
| 2. Colágeno tipo 2 | 22. Displasias osteoscleróticas neonatales |
| 3. Colágeno tipo 11 | 23. Densidad ósea aumentada |
| 4. Trastornos de sulfatación | 24. Densidad ósea aumentada con compromiso meta y/o diafisiario |
| 5. Perlecan | 25. Osteogénesis imperfecta y densidad ósea disminuida |
| 6. Agrecan | 26. Mineralización defectuosa |
| 7. Filamina | 27. Enf. de depósito lisosomal con compromiso esquelético |
| 8. TPRV4 | 28. Osteolisis |
| 9. Displasias con costillas cortas (con o sin polidactilia) | 29. Desarrollo esquelético desorganizado |
| 10. Displasias epifisiaria múltiple y pseudoacondroplasia | 30. Síndromes con sobrecrecimiento y compromiso esquelético |
| 11. Displasias metafisiarias | 31. Osteoartropatías genéticas inflamatorias |
| 12. Displasias espondilo metafisiarias | 32. Displasia cleidocraneal y defectos de osificación |
| 13. Displasias espondiloepimetafisiarias | 33. Síndromes de craneosinostosis |
| 14. Displasias espondilodisplásicas severas | 34. Disostosis con compromiso craneofacial |
| 15. Displasias acromélicas | 35. Disostosis con compromiso vertebral y costal |
| 16. Displasia acromesomélica | 36. Disostosis rotulianas |
| 17. Displasias mesomélicas y rizomesomélicas | 37. Braquidactilias |
| 18. Displasias con “huesos curvos” | 38. Defectos de reducción-hipoplasia de extremidades |
| 19. Displasias con “huesos delgados” | 39. Polidactilia-sindactilia-trifalangismo |
| 20. Displasias con luxaciones articulares múltiples | 40. Defectos en la formación articular y sinostosis |
Al realizar el estudio genético-molecular de estas enfermedades, 316 presentaron alteraciones en alguno de los 226 genes descritos a esa fecha como relacionados con trastornos primarios del esqueleto.
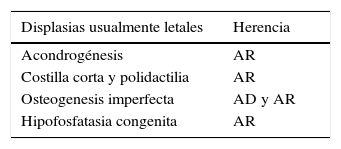
Las DE que se presentan y sospechan en forma precoz se acompañan por lo general de un compromiso más severo y se estima que una cuarta parte de ellas podrían ser letales en el período neonatal temprano (Tablas 2 y 3).
Sabemos que los pacientes con DE requieren de un manejo multidisciplinario e integral, que día a día conocemos de nuevos genes involucrados en la patogenia de estas enfermedades y que contamos con nuevas posibilidades terapéuticas para ofrecer a nuestros pacientes.
Es así que los autores de este capítulo consideramos importante partir haciendo un énfasis especial en la sospecha clínica temprana, destacar luego la necesidad de un estudio radiológico adecuado y en manos expertas y finalmente conocer que existen posibilidades terapéuticas que pueden mejorar deformidades y eventualmente la talla final. Estas técnicas quirúrgicas han tenido grandes avances en los últimos tiempos, especialmente en lo que se refiere al alargamiento óseo. Tablas 2 y 3.
SOSPECHA CLÍNICAUna adecuada aproximación clínica permitirá, tras una evaluación radiológica en manos expertas una orientación diagnóstica más precisa, que puede ser confirmada con un estudio molecular específico.
El tener un diagnóstico preciso es fundamental en decidir una adecuada conducta terapéutica, incluyendo un correcto asesoramiento al paciente y su familia.
La forma de presentación clínica que con mayor frecuencia conduce al pediatra o al especialista (endocrinólogo o genetista) hacia la sospecha de una DE es la talla baja.
Se define como talla baja aquella ubicada bajo el percentil 3 o bajo 2 desviaciones estándar de las curvas de referencia según edad, sexo y etnia.
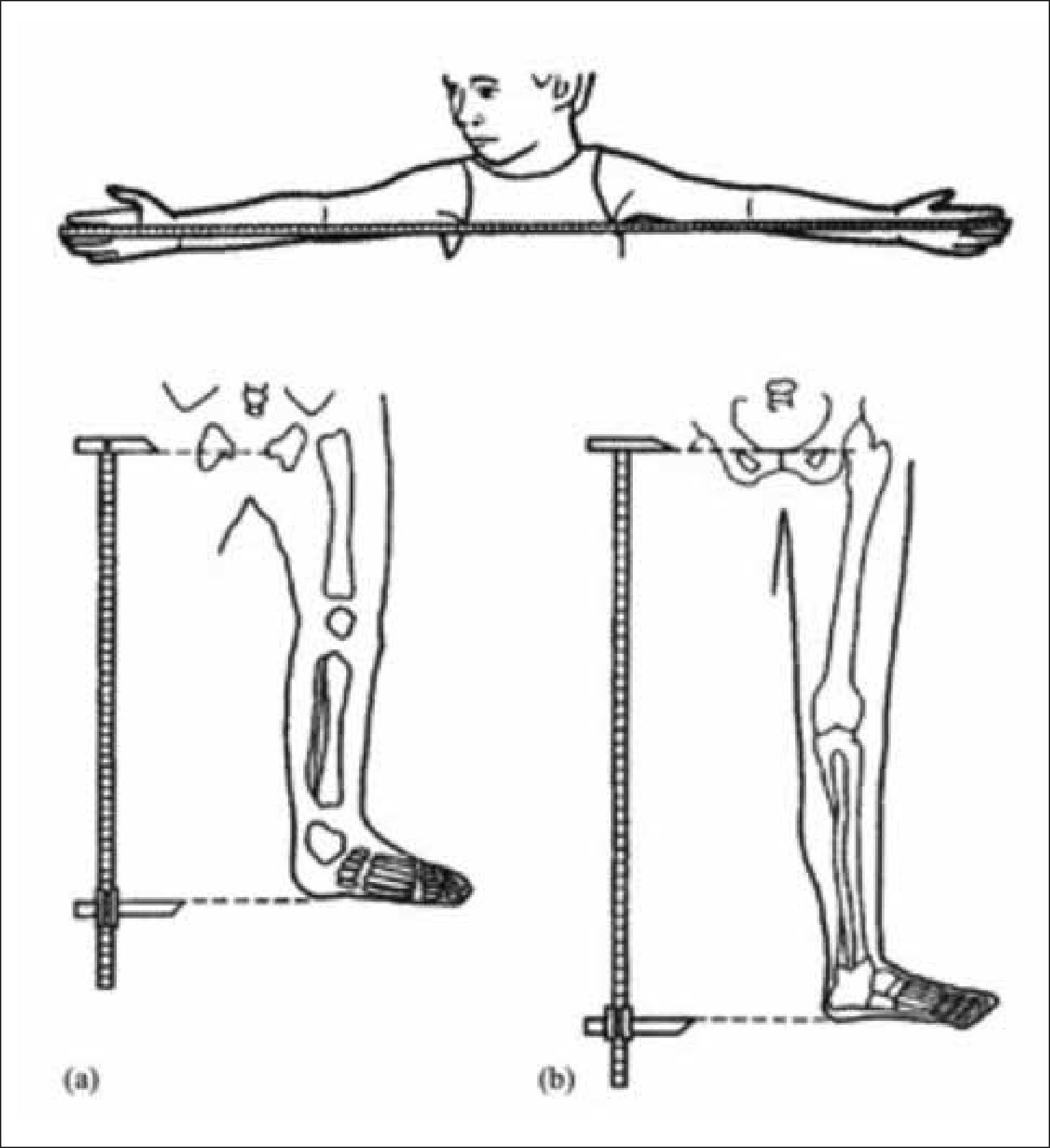
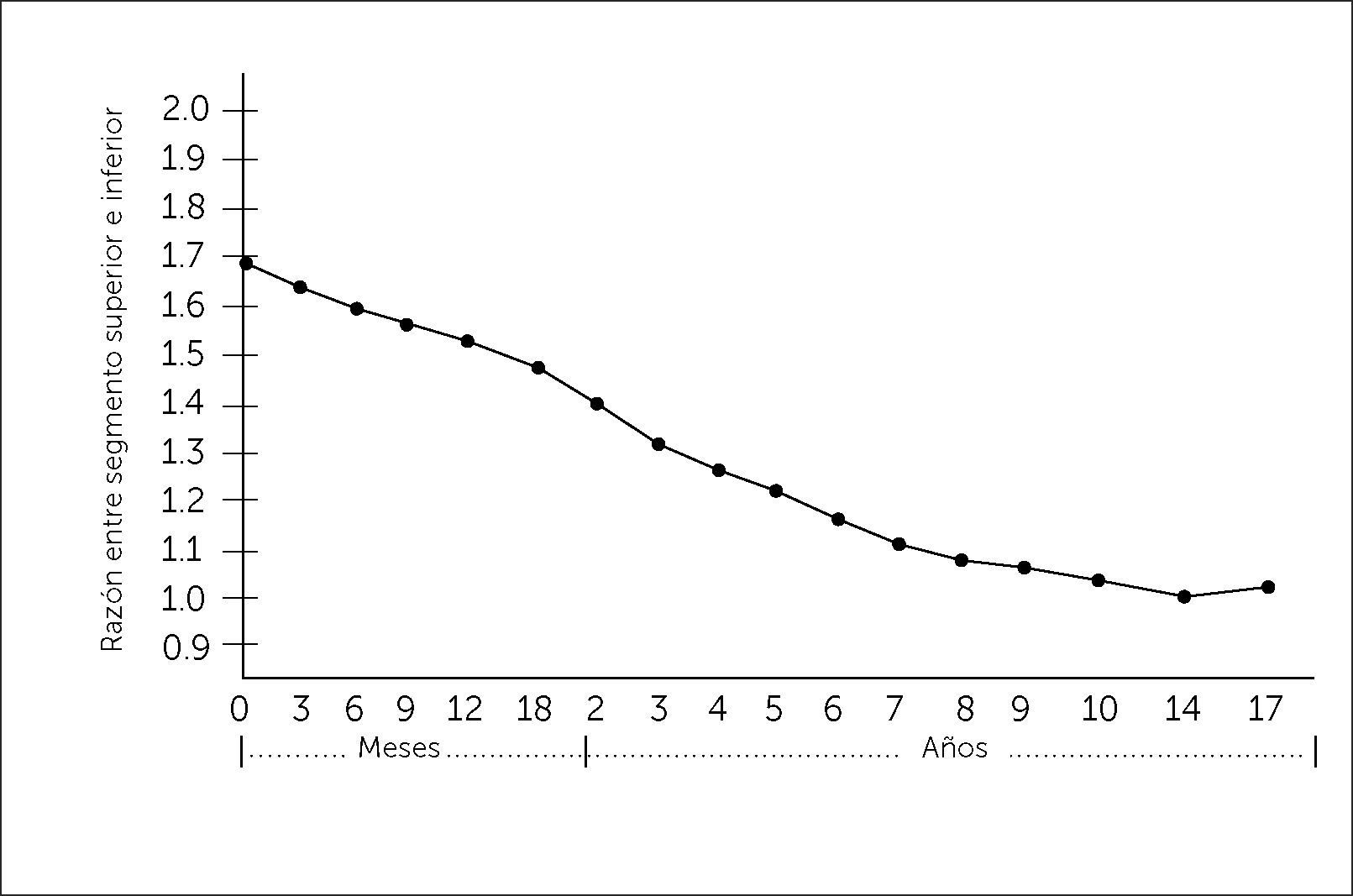
Al evaluar un paciente con talla baja, debemos cuidadosamente analizar las proporciones corporales. Estas deben incluir la medición del perímetro craneano, la envergadura o distancia entre la punta de los dedos medios de las manos, la medición del segmento inferior o distancia entre la sínfisis púbica (Figura 1) y la planta de los pies y el segmento superior, que se calcula con la diferencia entre la talla y el segmento inferior o se infiere de la medición de talla sentado. La desproporción corporal se objetivará tras comparar estas medidas con los valores de referencia normales por edad (Figura 2)3.


Luego de definirse que el paciente tiene talla baja desproporcionada se deberá especificar, si existe un acortamiento de extremidades, si este es proximal o que afecta el húmero y/o el fémur (compromiso rizomélico), medial o que afecta radio/cúbito y/o tibia/peroné (compromiso mesomélico) o si es distal o que afecta manos y/o pies (compromiso acromélico). También según las proporciones corporales podemos definir exista un acortamiento de tronco que nos hace sospechar clínicamente compromiso de columna4.
También debemos evaluar la presencia de malformaciones asociadas, ya sean menores y/o mayores (cardiacas, oculares u otras) y el desarrollo psicomotor, que pueden orientar a un diagnóstico específico.
En la historia clínica es fundamental la búsqueda de antecedentes familiares que orienten a DE, considerar talla de familiares, consanguinidad y/o el antecedente de abortos o mortinatos. Se debe preguntar por historia de osteoartritis precoz, prótesis de caderas, escoliosis grave, entre otras. Además, debemos realizar una genealogía que incluya, idealmente, al menos tres generaciones.
Muchas veces la talla baja es un diagnóstico que se repite en varios integrantes de una familia, así la talla baja familiar se ha considerado por mucho tiempo como una variante normal del crecimiento. Hoy, gracias a la posibilidad de estudios moleculares, hemos comprendido que muchas de estas tallas bajas familiares pueden corresponder a una DE y que los afectados podrían beneficiarse de alguna terapia específica.
Cuando contamos con una historia familiar positiva para una determinada DE, podemos prospectivamente sospechar afección en periodo prenatal o frente a ciertos hallazgos ecográficos. Dentro de estos se incluye la presencia de retraso del crecimiento intrauterino, desproporción de segmentos, alteraciones de la densidad ósea y presencia de fracturas. Frente a algunas asociaciones, ejemplo: fémur corto, frente abombada, puente nasal plano, manos en tridente, podemos sospechar una acondroplasia. Frente al hallazgo de fracturas en periodo intrauterino, lo más probable que corresponda a una ontogénesis imperfecta.
Es importante intentar establecer un diagnóstico lo más preciso posible en el prenatal, especialmente ante la sospecha de una DE letal, incluyendo incluso estudios genéticos específicos. Esto permitirá planificar el manejo obstétrico y neonatal y realizar un adecuado asesoramiento genético a la familia6.
Luego, los endocrinólogos destacamos la necesidad de un control pediátrico periódico que permita realizar una curva de crecimiento desde el periodo neonatal. Esta curva junto a los antecedentes familiares y la medición de las proporciones corporales permitirán en forma más clara sospechar patología esquelética subyacente.
Las DE son un desafío diagnóstico complejo, se requiere de una alta y temprana sospecha clínica, incluso desde el prenatal, del apoyo de especialistas expertos que trabajen en forma coordinada permitiendo así ofrecer eventuales terapias en forma oportuna y un apoyo global a la familia.
ESTUDIO IMAGENOLÓGICO EN SOSPECHA DE DISPLASIA ESQUELÉTICALa evaluación por imágenes en estos pacientes se orienta a confirmar o descartar la sospecha clínica de DE u otra alteración ósea.
Habitualmente el estudio radiológico se solicita en niños con talla baja o pacientes que nacen con alguna malformación o dismorfias, en que se sospecha un compromiso radiológico subyacente7.
Existen variadas guías y recomendaciones que intentan definir los exámenes radiológicos requeridos en el estudio de los niños con sospecha de DE8,9.
Los pacientes que clínicamente se evalúan como “baja estatura proporcionada”, sin otras alteraciones al examen físico, sólo requieren una radiografía de mano para evaluación de edad ósea como estudio inicial por imágenes, evitando así exposición innecesaria a radiación10.
En pacientes con “talla baja desproporcionada” se recomienda el estudio radiológico genético, también llamado estudio esquelético, que consiste en radiografías de los distintos segmentos corporales que pueden aportar al diagnóstico definitivo, considerando siempre involucrar la menor dosis de radiación para el niño.
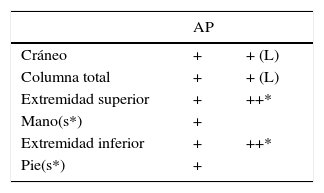
En la Tabla 4 se pueden observar las proyecciones recomendadas, tanto para los pacientes que han sido considerados simétricos como aquellos asimétricos; este estudio considera el mínimo de radiografías que permiten evaluar el esqueleto completo.
Segmentos óseos que deben incluirse en el estudio esquelético, considerando la menor exposición a radiación; pelvis y tórax se evalúan en Rx columna total AP
| AP | ||
|---|---|---|
| Cráneo | + | + (L) |
| Columna total | + | + (L) |
| Extremidad superior | + | ++* |
| Mano(s*) | + | |
| Extremidad inferior | + | ++* |
| Pie(s*) | + |
* En caso de asimetría, se consideran Rx de huesos largos de ambos lados y ambas manos/pies.
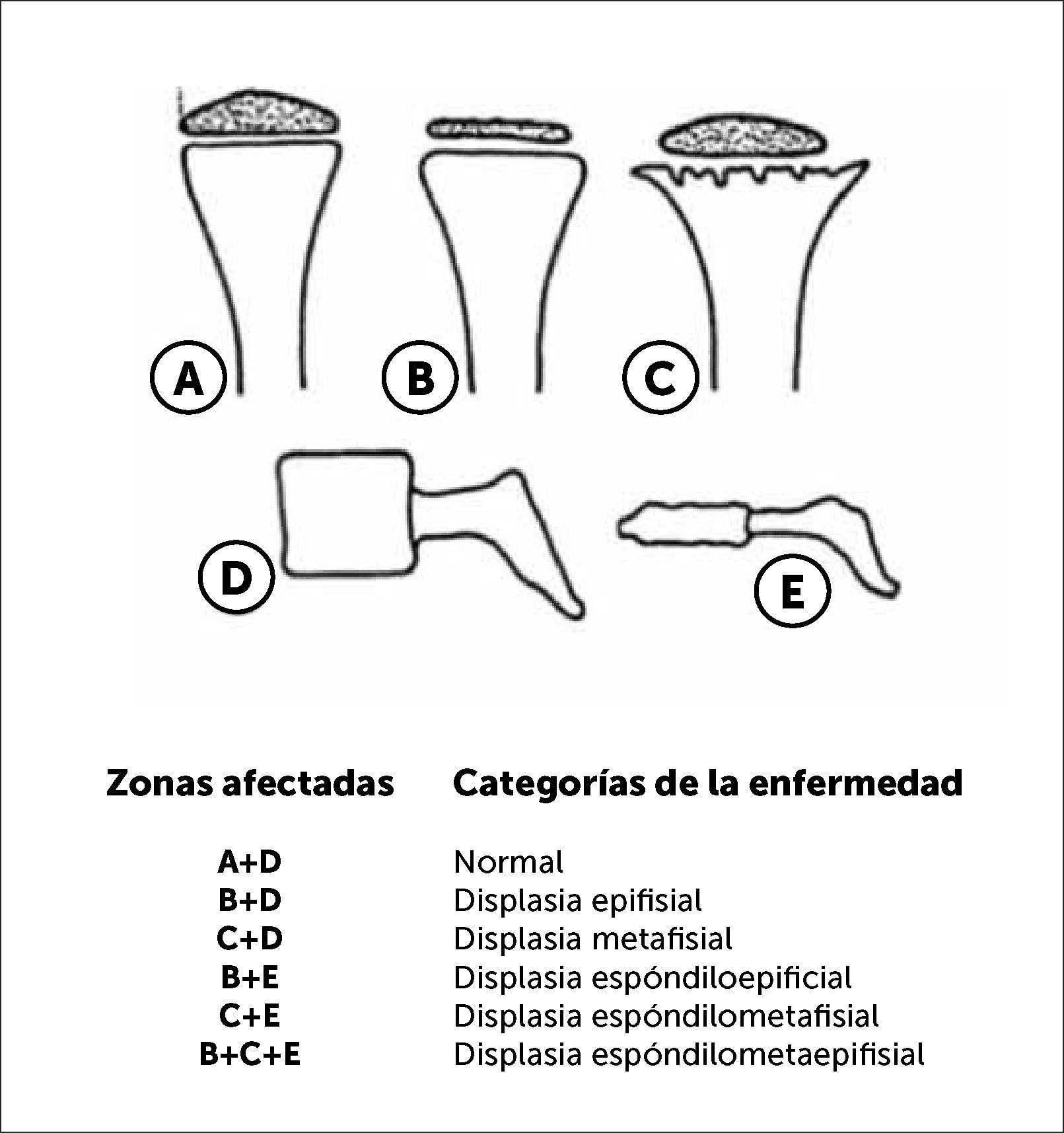
En la Figura 3 podemos observar una clasificación práctica y útil en la orientación diagnóstica según segmento óseo comprometido, epifisiario, metafisiario, diafisiario y de columna (espondilo).

Describiremos brevemente la importancia de los segmentos incluidos en el estudio esquelético y el significado de los hallazgos más frecuentes:
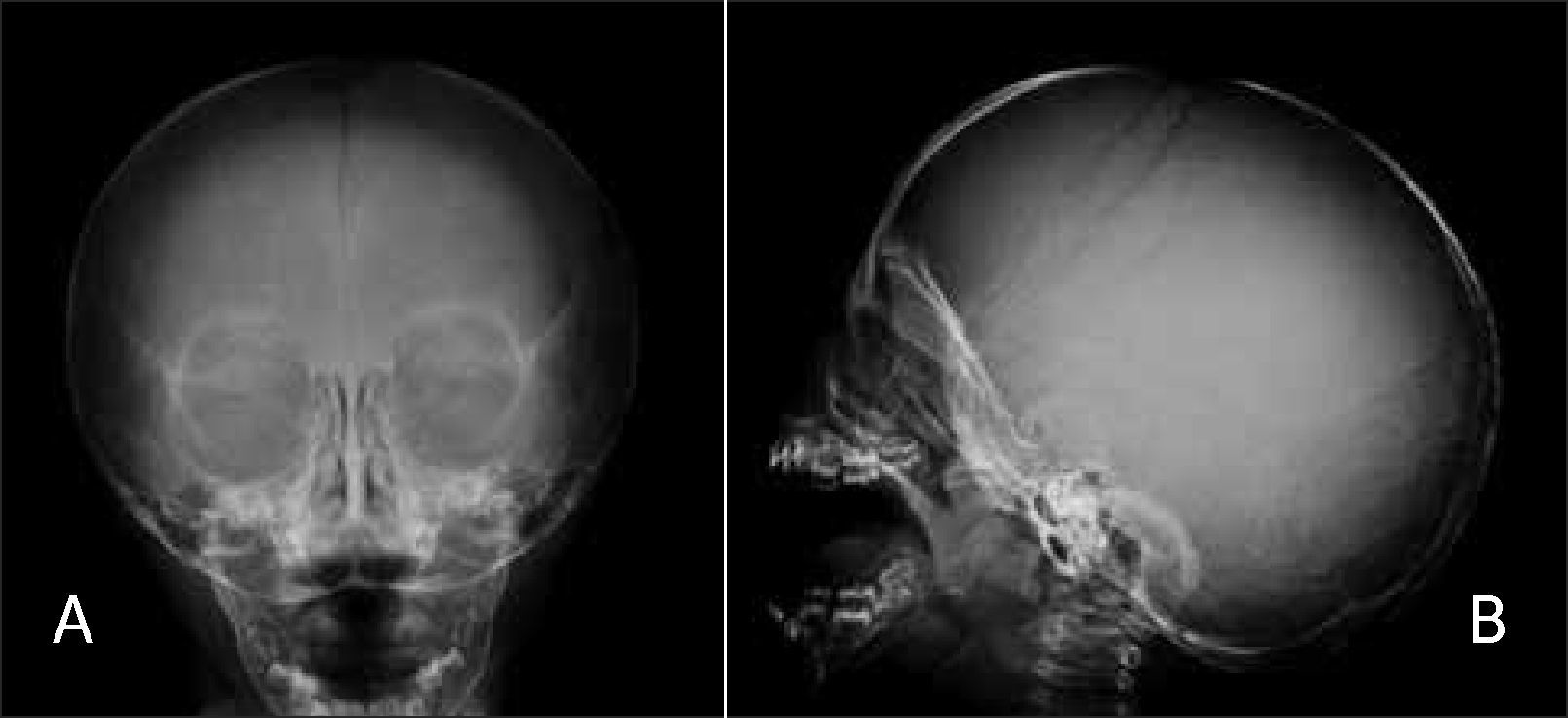
1.1CráneoLa radiografía de cráneo puede proporcionar información adicional para plantear o apoyar una hipótesis diagnóstica. El examen radiológico puede confirmar la sospecha clínica de micro/macrocefalia, demostrar alteraciones en la osificación o presencia de craneosinostosis, hallazgos que pueden estar presentes en diversos cuadros patológicos. Entre otros, en la displasia cleidocraneal típicamente se observa retardo en el cierre fontanelar con presencia de fontanela de gran tamaño, en osteogénesis imperfecta se observa alteración de osificación asociada a múltiples huesos wormianos (Figura 4) y los síndromes de Crouzon y Apert presentan craneosinostosis.
1.2Columna y costillas Rx Frontal, b) Rx Lateral.")
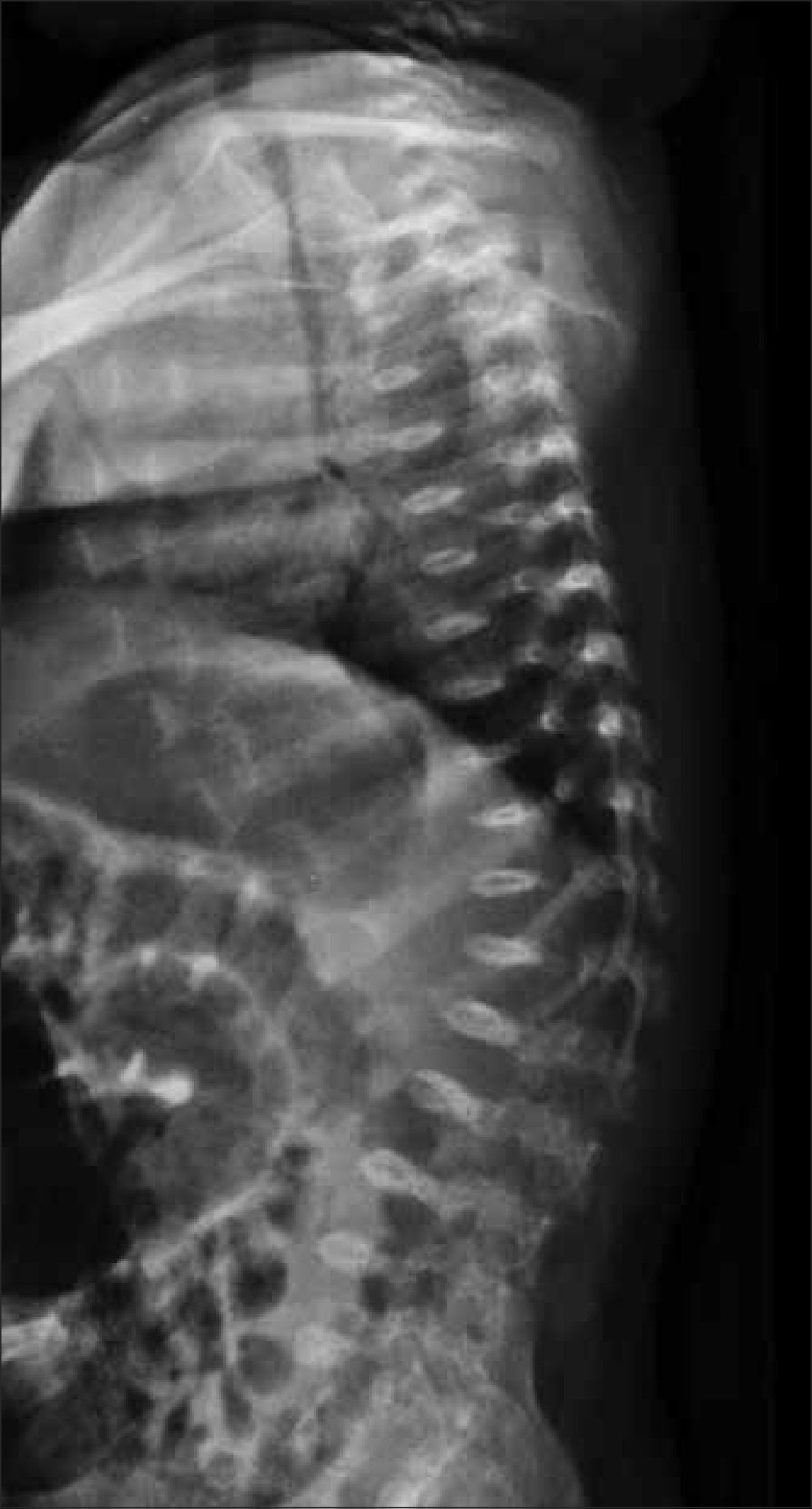
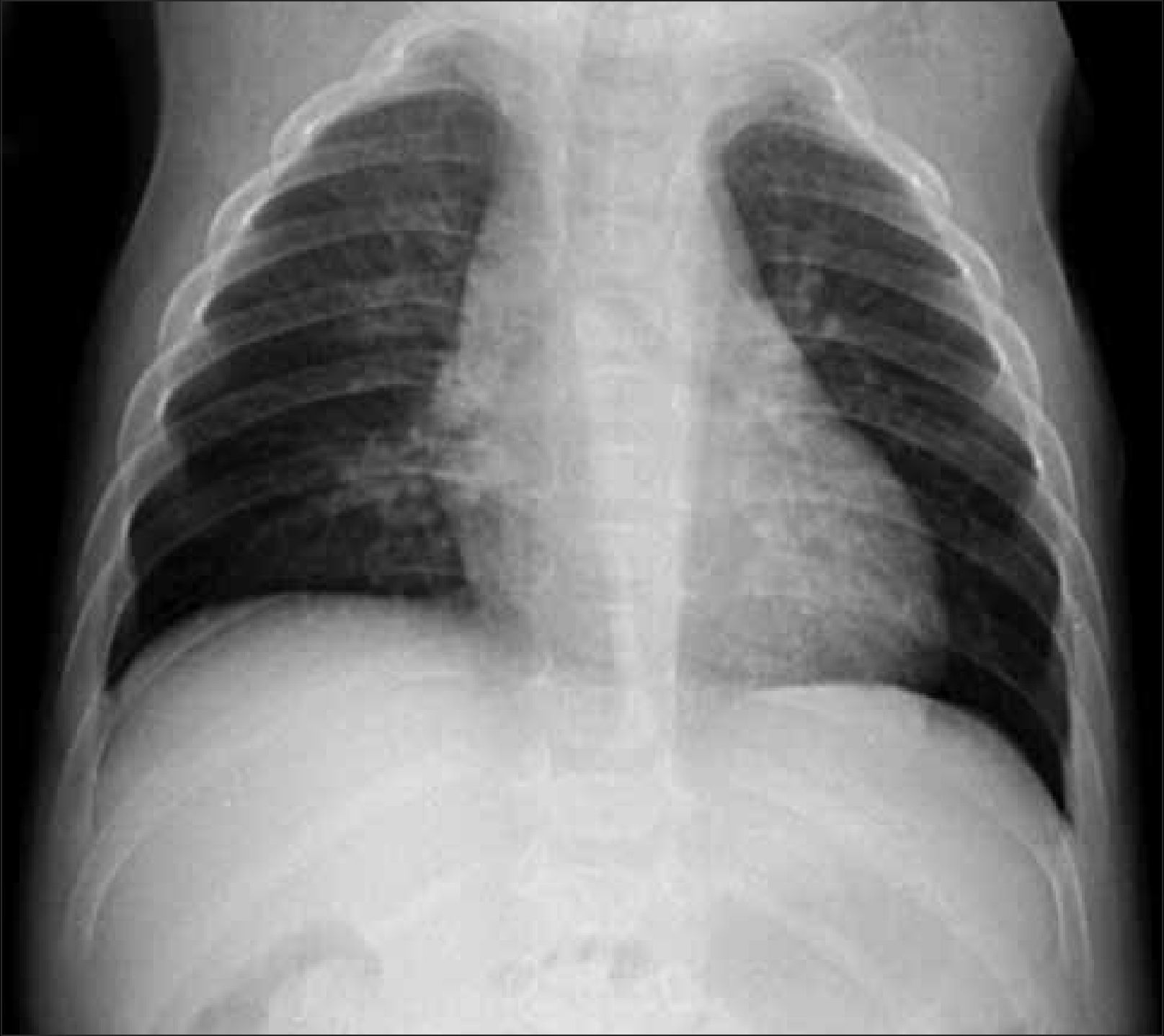
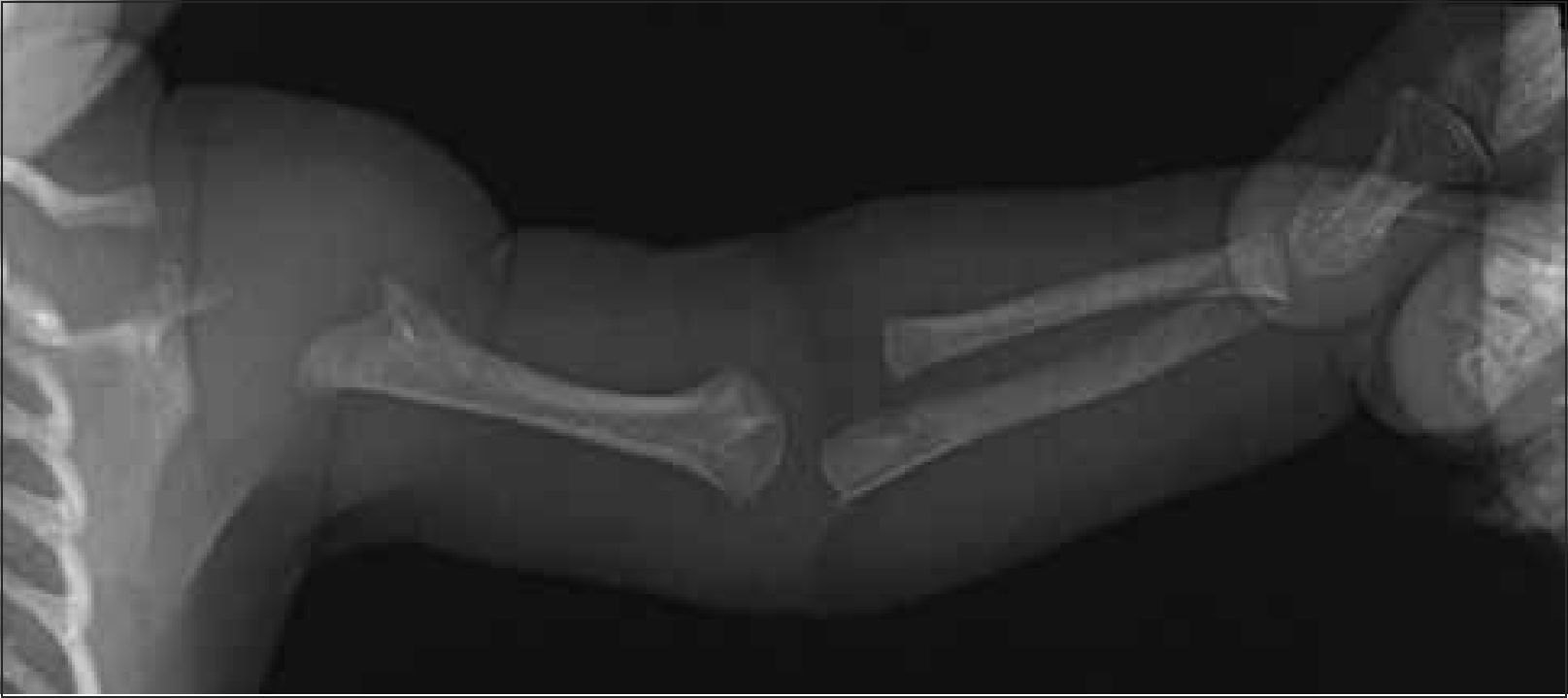
Se debe obtener radiografía de columna completa, en ambas proyecciones, incluyendo las vértebras cervicales. Debe observarse la forma y altura de los cuerpos vertebrales. El hallazgo de platiespondilia (vértebra aplanada) es de gran importancia diagnóstica ya que se observa en variados síndromes genéticos (Figura 5) como osteogénesis imperfecta, displasia espondilometafisiara o espondiloepifisiaria, entre otros. En el tórax, habitualmente incluido en la radiografía frontal de columna, es importante analizar dirigidamente las costillas, escápulas y clavículas (Figura 6).


Es necesario evaluar la amplitud del canal raquídeo y medir la distancia interpedicular lumbar baja en radiografía frontal, cuyo estrechamiento progresivo a nivel lumbosacro puede indicar la presencia de acondroplasia o hipocondroplasia (Figura 7).
1.3Pelvis
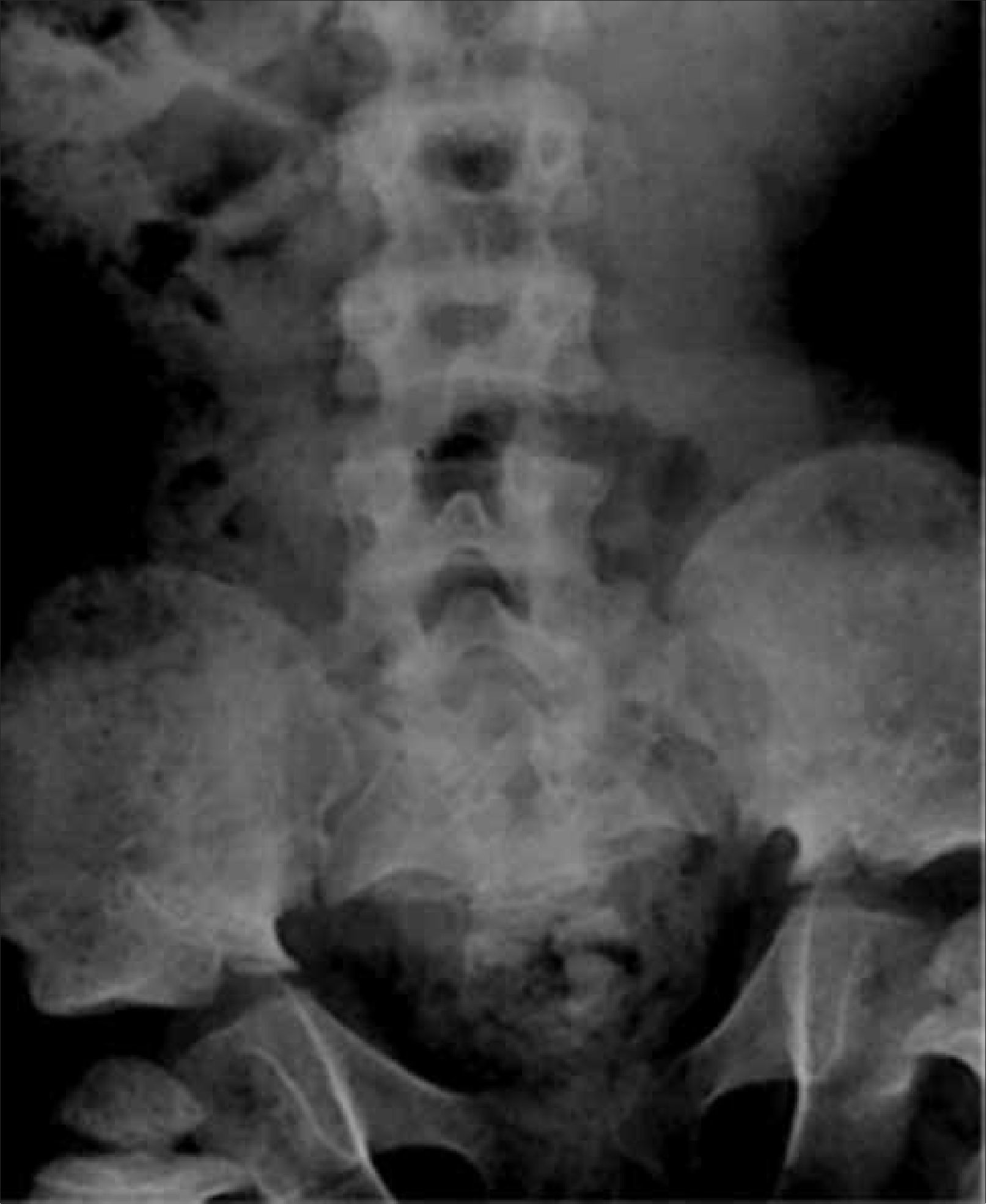
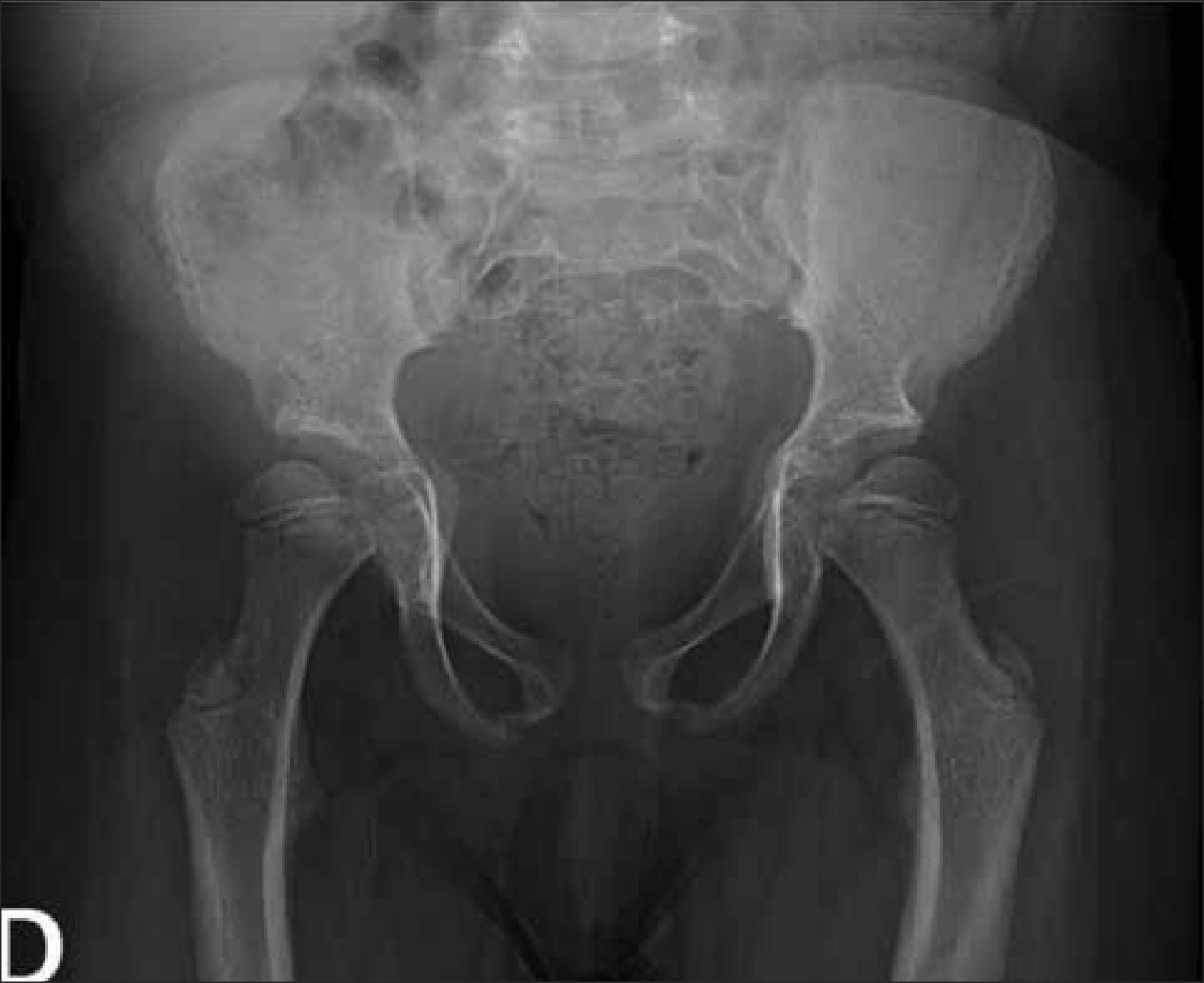
Este segmento también puede ser incluido en Rx columna, evitando exposición adicional a radiación. Es necesario observar forma y tamaño de las alas ilíacas, además del grado de osificación del pubis, hallazgos que orientan hacia algunas displasias esqueléticas. En DE las alteraciones observadas a nivel de pelvis son muy variadas; a modo de ejemplo, la displasia cleidocraneal presenta retardo de osificación del pubis, las displasias metafisiarias precozmente evidencian alteración metafisiaria asociada a coxa vara y las mucopolisacaridosis muestran habitualmente alas ilíacas pequeñas, subluxación de cabezas femorales y coxa valga (Figura 8).
1.4Huesos largos
Resulta extremadamente importante hacer un análisis detallado de las mediciones de los distintos segmentos óseos para diferenciar si el acortamiento es:
Rizomélico, del aspecto proximal de extremidades superiores/inferiores (Figura 9), hallazgo típicamente observado en acondroplasia e hipocondroplasia.

Mesomélico, del segmento medio o bajo de extremidades superiores/inferiores. Se observa en variadas DE, entre ellas la discondreosteosis de Leri Weill y el síndrome de Ellis van Creveld.
Acromélico, de manos y/o pies, observado en displasias acromélicas y acromesomélicas.
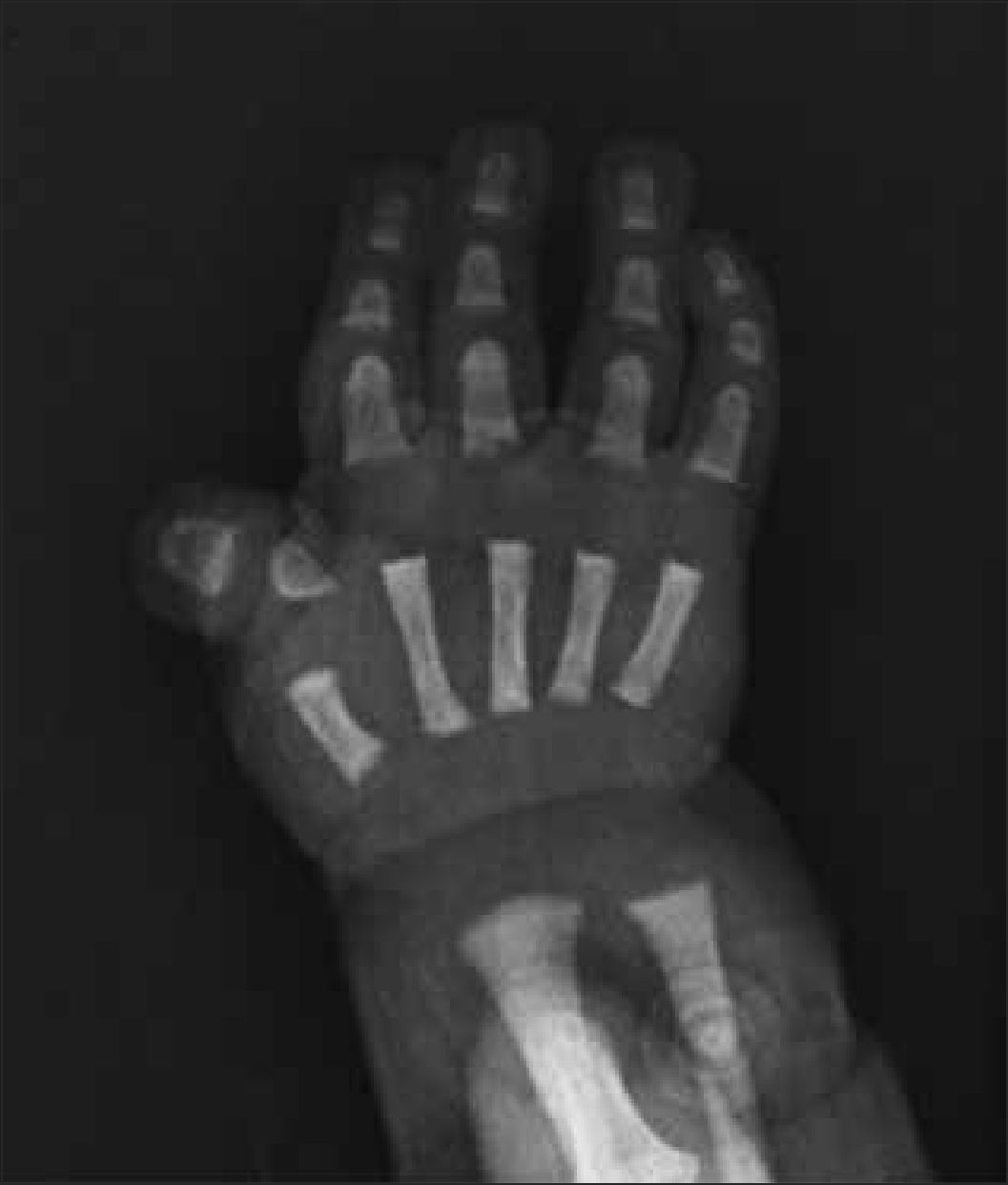
1.5Manos y piesHabitualmente se incluye la radiografía de mano para evaluación de la edad ósea, como evidencia de la maduración del esqueleto. Las anomalías morfológicas de las manos son muy frecuentes y variadas en las DE, por esto las radiografías deben ser observadas con detalle, incluyendo todos los segmentos óseos de la mano y las partes blandas, que pueden evidenciar por ejemplo, sindactilias cutáneas.
Existen múltiples hallazgos que orientan el diagnóstico, como por ejemplo, alteraciones metafisiarias o epifisiarias, dedos en tridente en acondroplasia, sindactilias en síndrome de Apert, alteraciones de ortejos y pulgares en síndrome de Rubinstein Taybi (Figura 10), entre muchos otros.

Los pies contribuyen poco al diagnóstico de DE, aunque muchas veces es posible encontrar anormalidades semejantes a las observadas en la manos.
En los pacientes sin hallazgos específicos se recomienda repetir el estudio después de dos años, especialmente si el niño continúa en un bajo carril de crecimiento. Sabemos que muchas alteraciones se hacen más evidentes en el tiempo. De esto deriva también, la posibilidad de estudio de padres u otros familiares afectados, al enfrentarnos a un niño pequeño con sospecha clínica de DE.
La dosis de radiación involucrada en los estudios esqueléticos acotados, efectuados con los equipos disponibles en la actualidad, puede ser considerada baja por lo que el estudio radiológico sigue siendo una herramienta diagnóstica útil y segura en el estudio de pacientes con sospecha de DE, especialmente niños con talla baja desproporcionada8.
RECONSTRUCCIÓN DE EXTREMIDADES EN DISPLASIAS ESQUELÉTICASDescribiremos en qué consiste el manejo quirúrgico ortopédico de los pacientes portadores de DE.
Las DE son un grupo muy heterogéneo de enfermedades y el enfrentamiento quirúrgico de ellas apunta a corregir básicamente tres problemas comunes: deformidades de las extremidades, desproporción y talla baja11.
La técnica a utilizar, el timing y las alternativas de fijación del hueso van a depender en gran medida del tipo de displasia, el pronóstico de talla final y la edad del paciente. Asimismo, es muy importante tener en consideración las expectativas del paciente y sus padres al momento de plantear una intervención de esta naturaleza.
1.6Consideraciones anatómicasLos pacientes con DE suelen tener huesos de menor tamaño (longitud y/o diámetro) que lo correspondiente para su edad12. La mayoría presenta deformidades de distinta magnitud en los huesos largos. La relación de longitud, entre los segmentos proximal y distal de las extremidades también suele estar alterada12–14. Finalmente, la densidad mineral ósea también puede ser anormal: desde huesos muy frágiles (osteogénesis imperfecta) hasta huesos extremadamente duros (piknodisostosis o meloreostosis)15,16.
Todos estos factores deben ser considerados al momento de planificar una intervención reconstructiva, de manera de elegir el tipo de osteotomía, la ubicación de ésta, los implantes a utilizar y el protocolo de rehabilitación a seguir.
1.7Consideraciones etiológicasCada grupo de DE poseen algunas características distintivas a considerar.
En primer lugar, debemos mencionar aquellas en las que existe compromiso de la columna vertebral. Aunque existen experiencias de alargamiento progresivo de columna mediante fijación externa en animales (perros y ovejas) sin ocasionar problemas neurológicos, en humanos no es planteable, por lo que en este grupo no es recomendable un alargamiento de las extremidades, pues aumentaría la desproporción11,17–19.
En DE con compromiso epifisiario, el proceso degenerativo del cartílago articular está acelerado. Estos pacientes habitualmente desarrollan precozmente artrosis de caderas y rodillas20–24. Durante el proceso de distracción ósea que se utiliza para alargamiento de las extremidades, se generan fuerzas importantes de compresión en el cartílago articular, que pueden acelerar aún más la destrucción de éste. Por tal motivo, en estos pacientes es preferible realizar alargamientos de poca magnitud (máximo 5cm) en varias oportunidades, en vez de grandes alargamientos25. Además, desde un punto de vista técnico, es recomendable “puentear” las articulaciones, de manera de protegerlas de estas fuerzas compresivas.
1.8Consideraciones quirúrgicasPara el cirujano ortopédico en DE, la planificación preoperatoria es la piedra angular para lograr el éxito del tratamiento. Los factores que se deben considerar incluyen la edad del paciente, la etiología, el o los huesos comprometidos, la presencia de deformidades y la localización de estas, la talla y el pronóstico de talla final, los implantes a utilizar, el número de cirugías, las expectativas de la familia, y finalmente, no podemos olvidar la situación económica de la familia, pues como podemos imaginarnos, estas cirugías requieren tecnología sofisticada, hospitalización en centros de alta complejidad y un seguimiento y rehabilitación prolongado.
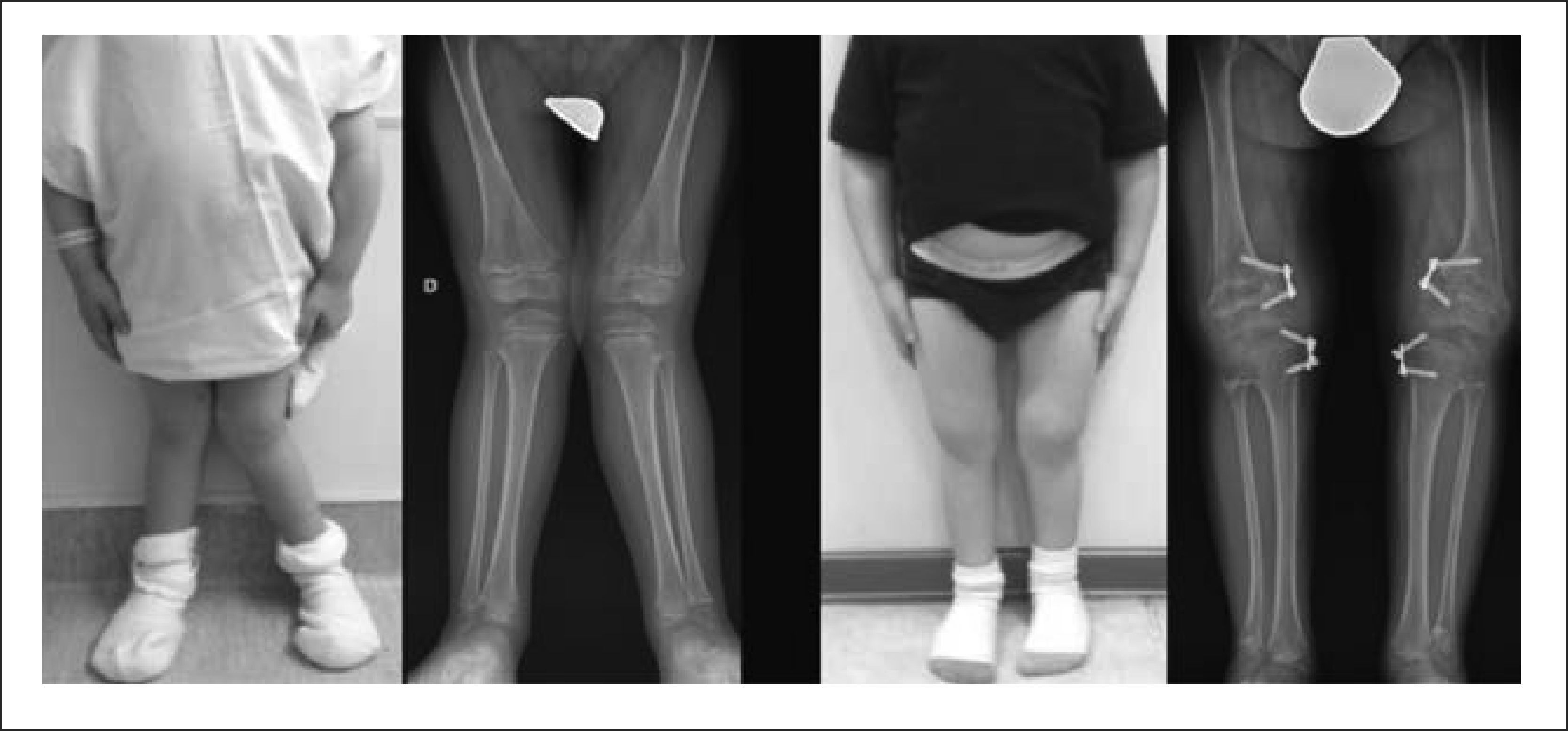
1.8.1a. EdadEs un factor importantísimo al momento de plantear una cirugía reconstructiva. El esqueleto infantil presenta cartílagos de crecimiento (fisis), lo que le confiere una capacidad de remodelación mayor que el esqueleto del adulto. Mediante técnicas mínimamente invasivas, es posible modular el crecimiento del cartílago, permitiendo correcciones angulares de los huesos largos en forma progresiva27–30. Esta técnica, también llamada Hemiepifisiodesis (Hemi: Mitad, Epifisio: Epífisis, Desis: Fusión), se puede emplear en varias condiciones en que las deformidades tienen su ápex cerca del cartílago de crecimiento (Figura 11). Con los implantes más modernos, es posible frenar en forma transitoria el crecimiento de una parte de la fisis, de manera que, una vez corregida la deformidad, el implante se retira y el hueso sigue creciendo en forma normal.

Como el compromiso suele ser bilateral, y esta técnica es poco invasiva, realizamos la hemiepifisiodesis bilateral en forma simultánea.
Cuando las deformidades son mayores, o se encuentran alejadas de la fisis, es necesario hacer osteotomías. En este sentido, el esqueleto infantil ofrece la ventaja de una rápida consolidación, y una menor necesidad de utilizar grandes implantes.
1.8.2b. EtiologíaDependiendo del diagnóstico específico de la DE, las alternativas quirúrgicas pueden variar. Ya se mencionaron los casos con compromiso vertebral y con compromiso de las epífisis. Otro aspecto a considerar es la situación de las partes blandas. Algunas DE, en especial la Acondroplasia, se presentan con una disminución de la elasticidad de los tejidos peri articulares, que pueden afectar en forma negativa el resultado de las osteotomías de realineamiento o de alargamiento. Así es, como en estos pacientes, al momento de realizar correcciones óseas se debe alargar o liberar las partes blandas. Específicamente, para el alargamiento de fémur, es fundamental seccionar el tracto iliotibial. Para la corrección de la deformidad en varo de la tibia, es recomendable descomprimir en nervio peroneo, y para alargar la tibia, es necesario alargar el tendón de Aquiles31–33.
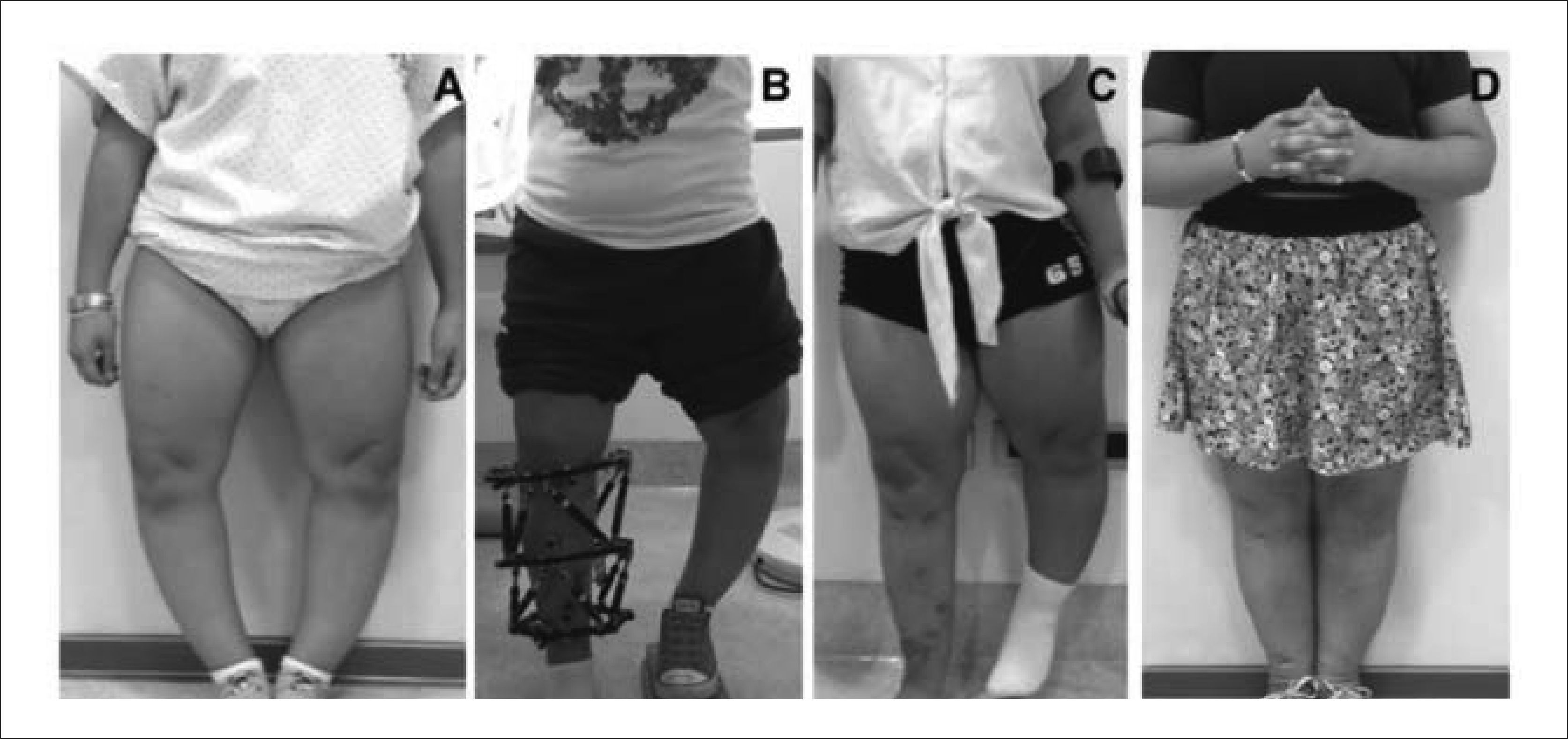
1.8.3c. Deformidades óseasEn la mayoría de las DE, las deformidades afectan ambas extremidades superiores y/o inferiores, ya sea en forma simétrica o siendo un lado más grave que el otro. Nuestra aproximación en estos casos, es corregir en una primera etapa un lado, de manera de permitirle al paciente usar la extremidad no operada para poder cargar su peso asistido por bastones. Esto le da mayor independencia al paciente, y facilita enormemente su rehabilitación y su reintegración sus actividades. En una segunda etapa, y una vez consolidados los huesos, se plantea la corrección de la otra extremidad. En la práctica, preferimos corregir simultáneamente fémur y tibia cuando existen deformidades en ambos huesos (Figura 12).
 Aspecto clínico pre operatorio. Nótese el marcado Genu Varo. B) Durante la corrección. C) Al termino de la corrección de la extremidad inferior derecha. D) Resultado final, luego de corrección bilateral. Publicación autorizada por paciente.")
RECONSTRUCCIÓN SECUENCIAL DE EXTREMIDADES INFERIORES EN RAQUITISMO FAMILIAR HIPOFOSFEMICO
A) Aspecto clínico pre operatorio. Nótese el marcado Genu Varo. B) Durante la corrección. C) Al termino de la corrección de la extremidad inferior derecha. D) Resultado final, luego de corrección bilateral. Publicación autorizada por paciente.
En la corrección de deformidades y alargamiento de huesos largos, se debe tener cuidado al momento de elegir el implante a utilizar para la fijación ósea. En el arsenal ortopédico, existen distintos tipos de fijación ósea, que incluyen placas, tornillos, clavos intramedulares, fijadores externos circulares, fijadores monolaterales, o combinaciones de éstos.
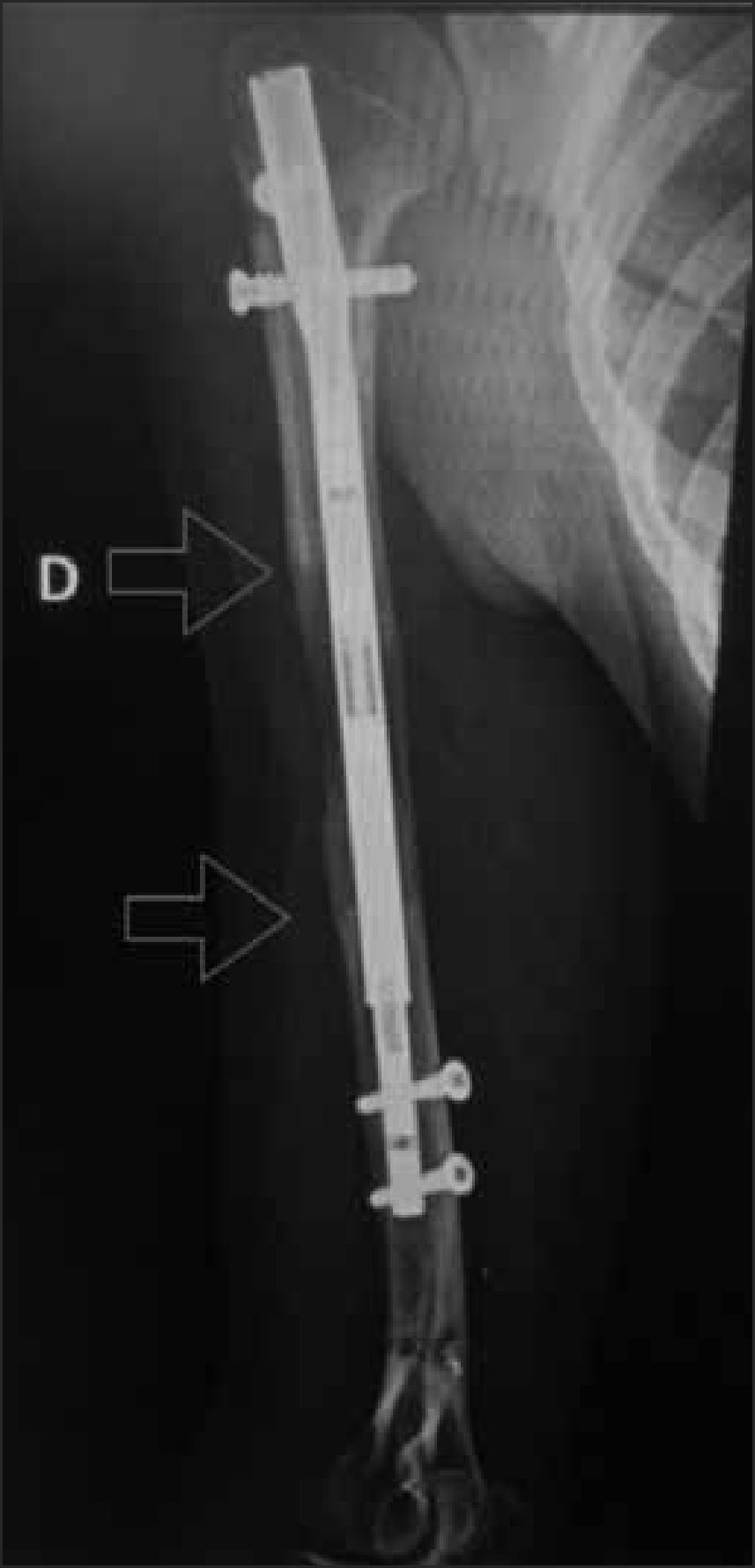
El implante ideal debiera ser aquel que brinde la fijación más estable, utilizando la técnica menos invasiva, que le brinde mayor comodidad al paciente y con la menor tasa de complicaciones. Desde el punto de vista biomecánico, los clavos intramedulares presentan una ventaja, pues permiten una carga precoz de la extremidad, se insertan a través de pequeñas incisiones, y habitualmente no alteran en forma significativa la irrigación del hueso, lo que se traduce en una consolidación más rápida. Además, brindan una mayor comodidad al paciente34–37 (Figura 13). Sin embargo, no todos los pacientes pueden ser candidatos a una fijación con este implante. En niños, al haber cartílago de crecimiento activo en los extremos del hueso, existe el riesgo de producir una lesión de éstos, que puede llevar a una detención del crecimiento. En algunas DE con esclerosis ósea, la inserción del clavo puede ser muy difícil, y en estos casos es mejor recurrir a otros implantes.
 Pre operatorio. B) Post corrección. Publicación autorizada por paciente.")
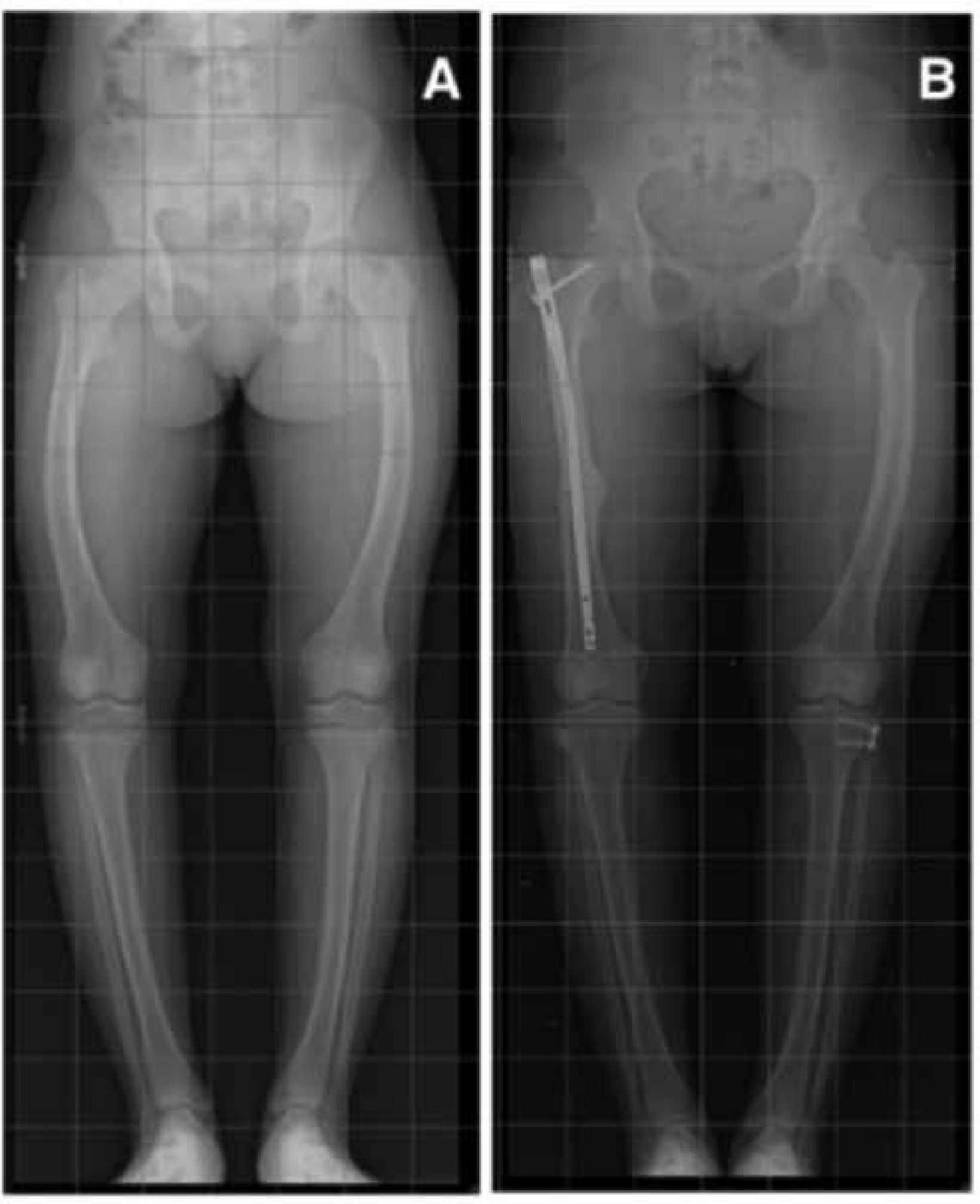
En los últimos años, una nueva generación de clavos intramedulares ha revolucionado el tratamiento de las deformidades óseas asociadas a acortamiento del segmento. Se trata de clavos expansibles, que requieren de abordajes mínimamente invasivos, que tienen la ventaja de alargarse, permitiendo estirar los
huesos afectados hasta 8cm sin necesidad de utilizar un fijador externo38–43. Estos dispositivos, se manejan a través de un control remoto, que se coloca sobre la piel. Entre los más conocidos y probados están el FitBone (Alemania), y el PRECICE (EE.UU.) (Figura 14). Este último es el único aprobado por la FDA.

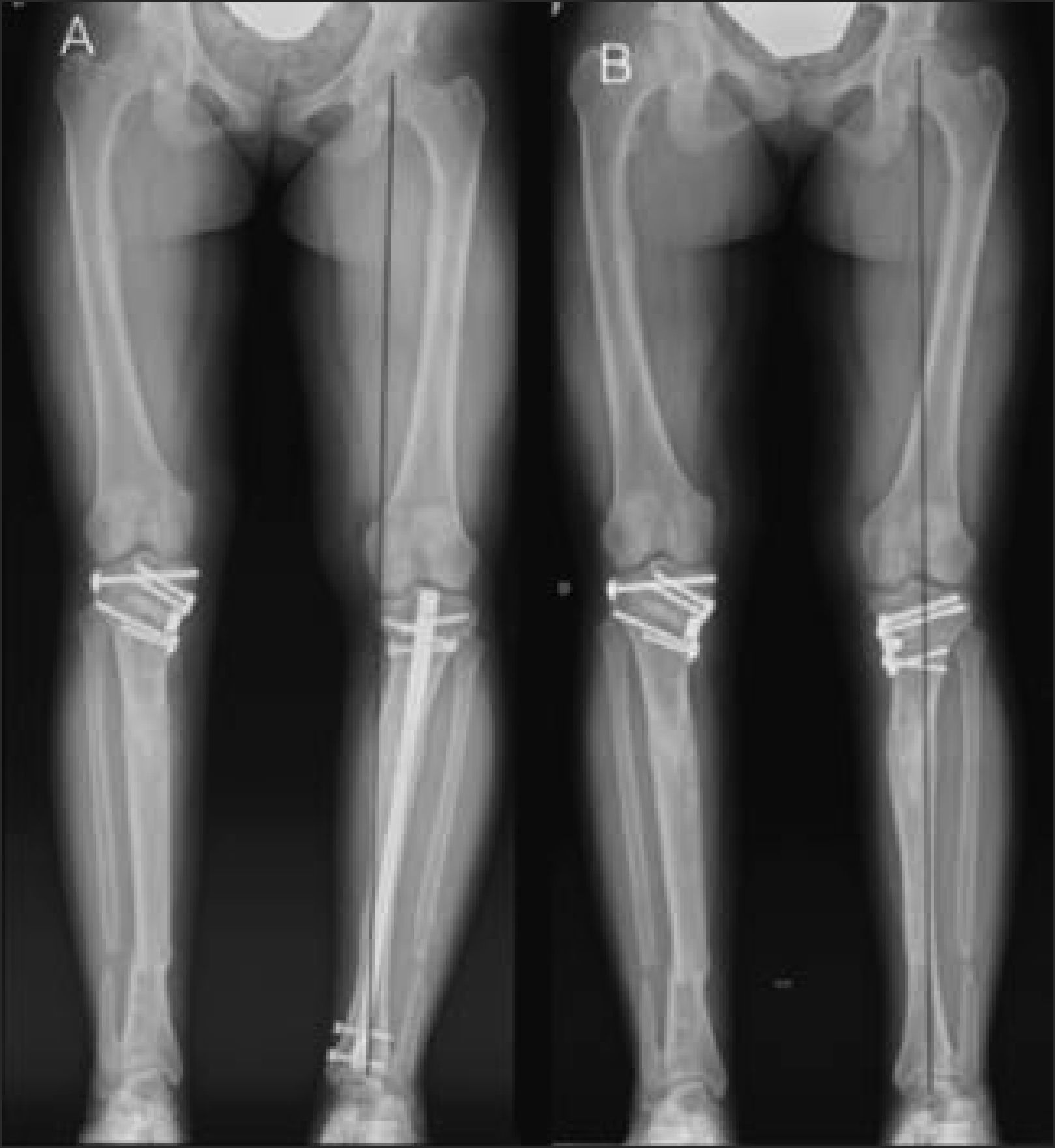
Las placas de osteosíntesis se utilizan de preferencia en adolescentes o adultos, que presentan deformidades simples y en un plano del espacio (ej. Varo o valgo, antecurvatum o recurvatum), y cuyo vértice se encuentra cerca de los extremos proximal o distal del hueso (Figura 15).
 En la Rx preoperatoria, se observa Genu Varo, a pesar de haberse intentado corrección con clavo intramedular. B) Se retiró el clavo, y se practicó una osteotomía valguizante y fijación con placa. Obsérvese que la línea del eje mecánico pasa por el centro de cadera, rodilla y tobillo.")
Corrección de Genu Varo con placa tipo Puddu® en paciente portadora de Raquitismo familiar.
A) En la Rx preoperatoria, se observa Genu Varo, a pesar de haberse intentado corrección con clavo intramedular. B) Se retiró el clavo, y se practicó una osteotomía valguizante y fijación con placa. Obsérvese que la línea del eje mecánico pasa por el centro de cadera, rodilla y tobillo.
Para el tratamiento de deformidades óseas en niños, el dispositivo de preferencia es el tutor externo. Si bien es incómodo y voluminoso en ocasiones, permite correcciones precisas, a través de un abordaje mínimamente invasivo, sin alterar ni lesionar los cartílagos de crecimiento. Además, ofrece la ventaja de poder alargar los segmentos comprometidos, de manera de abordar el tema de la talla simultáneamente.
1.8.5e. Talla finalEl tratamiento de la talla baja propiamente tal, es sin duda el aspecto más controvertido en el manejo de las DE. Existen varias organizaciones de pacientes portadores de talla baja, quienes se oponen a esta alternativa, argumentando razones éticas, sociales y/o médicas. Dentro de sus argumentos, destacan el hecho de que el alargamiento óseo no mejora el cuadro original, de modo que un paciente portador de una DE, seguirá siendo portador de ésta, a pesar de que haya sido “alargado”11,12.
Otro tema a discutir es definir las expectativas del paciente y sus padres. Por una parte, están aquellos que buscan una talla “funcional”. Considerar que una persona sobre los 150cm está en condiciones de conducir un automóvil sin necesidad de adaptaciones especiales y puede alcanzar objetos en la altura promedio (estantes de cocina, botón de parada de buses, etc.). Por otra parte, están aquellos que pretenden alcanzar una estatura “normal”. Esta meta varía considerablemente dependiendo del país donde se realice o de la etnia a la que pertenezca el paciente. Así, un paciente que es de talla normal baja en los países escandinavos, puede ser absolutamente normal en centro América.
Se puede finalmente hacer un plan de tratamiento mediante alargamientos óseos sucesivos sin olvidar que:
- 1.
Si consideramos que el máximo alargamiento que se puede lograr es una fracción del tamaño original, entonces a mayor longitud original del hueso a alargar, mayor alargamiento se puede realizar.
- 2.
En los alargamientos óseos para manejo de la talla, se deben tratar simultáneamente ambas extremidades.
- 3.
En condiciones normales, el ritmo de distracción es de 1mm/día y se puede alargar con bastante seguridad hasta 8cm por segmento. Si alargamos simultáneamente fémur y tibia, podría alcanzarse los 16cm en total en un mismo período de tiempo.
- 4.
Cuando se utilizan fijadores externos para realizar el alargamiento óseo, a modo aproximado podemos decir que por cada cm alargado, el tutor debe quedar in situ por 35-40 días 34. Así, luego de alargar 5cm la tibia, el tutor debe permanecer instalado por 175 a 200 días.
- 5.
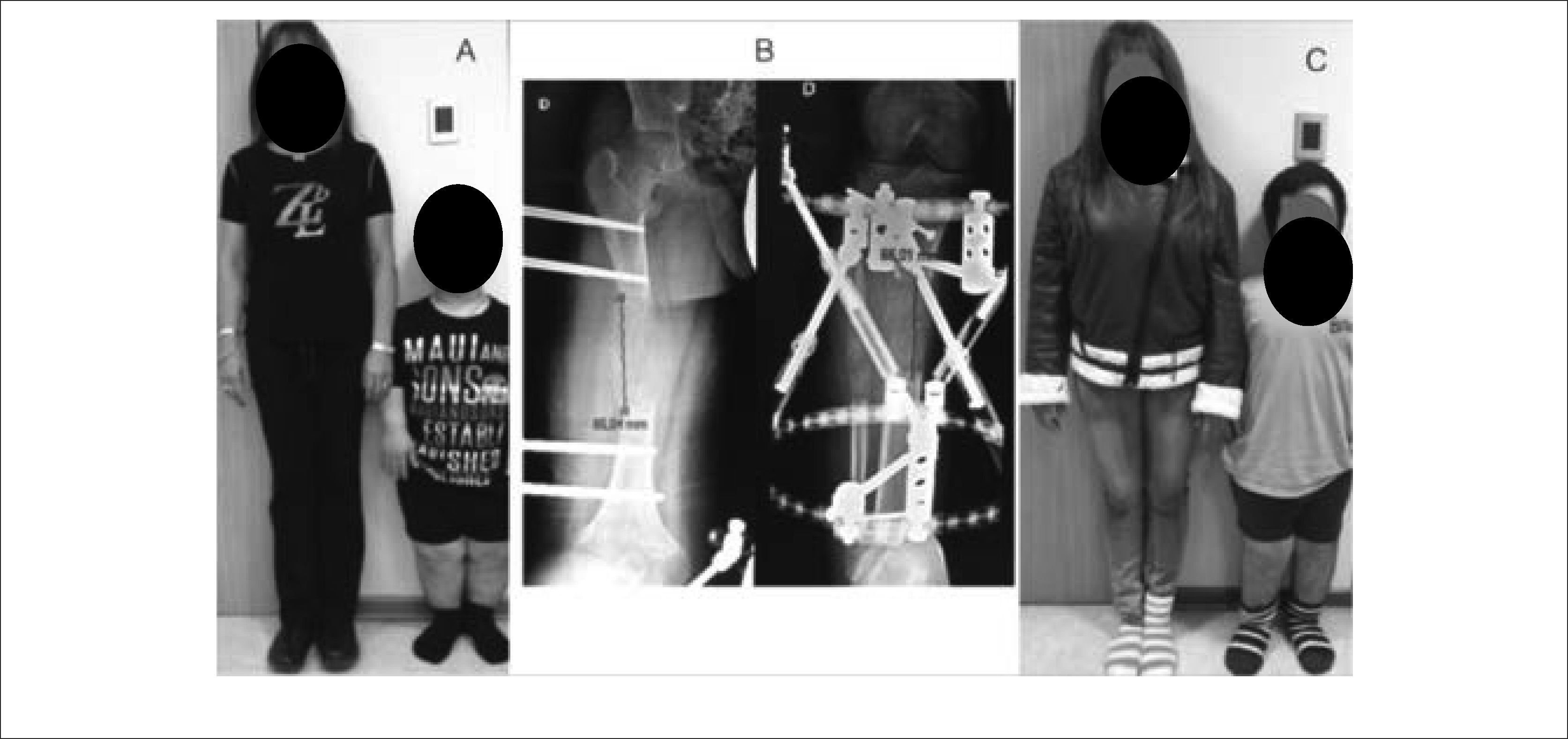
Cuando el pronóstico de talla final es muy limitado, se puede repetir el alargamiento óseo en distintas edades. Así por ejemplo, en pacientes portadores de Acondroplasia, pueden llevarse a cabo 4 procedimientos de alargamiento óseo para lograr una estatura “normal” 35. Se comienza a la edad de 7-10 años, de manera de alargar inicialmente 5cm por segmento (recordar que a menor longitud del hueso original, menor alargamiento), luego se repite a la edad de 13 años, donde ya es posible alargar 5-8cm por segmento. Nuevamente, a los 16 años se puede lograr 5-8cm por segmento, y finalmente a los 18-19 años, se puede hacer un alargamiento de fémur de 5-8cm adicionales (Figura 16). En este protocolo, también está contemplado el alargamiento de húmero, a los 14-15 años, que es bastante más simple en términos de incomodidad y de necesidad de reposo y otorga un beneficio funcional fundamental, pues estos pacientes habitualmente no tienen el alcance de brazos suficientes como para realizarse aseo en zona perineal. Luego de estos alargamientos, en situaciones ideales, el paciente podría aumentar su talla final en más de 40cm.
 Paciente junto a su madre preoperación. B) Se observa progresión de la distracción ósea en fémur derecho de aproximadamente 8cm y en tibia de 6cm. C) Resultado final. El paciente ha crecido 15cm. Publicación autorizada por paciente.") Figura 16.
Figura 16.Alargamiento de extremidades inferiores en Acondroplasia
A) Paciente junto a su madre preoperación. B) Se observa progresión de la distracción ósea en fémur derecho de aproximadamente 8cm y en tibia de 6cm. C) Resultado final. El paciente ha crecido 15cm.
Publicación autorizada por paciente.
(0.31MB).
 Paciente junto a su madre preoperación. B) Se observa progresión de la distracción ósea en fémur derecho de aproximadamente 8cm y en tibia de 6cm. C) Resultado final. El paciente ha crecido 15cm. Publicación autorizada por paciente.")
Es fundamental recalcar que el alargamiento de extremidades es un proceso largo, complejo y que requiere de mucho sacrificio por parte del paciente y sus padres. Durante la etapa de distracción, es necesario mantener al paciente en kinesiterapia en forma permanente, 5 veces por semana. No está en condiciones de caminar por un tiempo prolongado, por lo que debe desplazarse en silla de ruedas. Además, es necesario controlar periódicamente, con radiografías cada 15 días para ver el progreso de la regeneración ósea. A diferencia de otras intervenciones, aquí los resultados se observan al mediano plazo.
El éxito del tratamiento ortopédico depende tanto de la etiología específica como de la planificación cuidadosa, tomando en consideración múltiples factores. Sin duda, lo que más urge es la corrección de deformidades óseas que interfieren con la función normal del paciente, y que pueden llevar a artrosis precoz. El manejo de la talla es un tema que se discute constantemente en reuniones de especialistas y que tiene tanto defensores como detractores. Independientemente de esta controversia, el objetivo que debemos perseguir es darle al paciente la mejor funcionalidad con el menor sufrimiento posible. Y es en ese espíritu, que consideramos que el alargamiento óseo debiera llevar al paciente a una talla “funcional” y no necesariamente a una talla “normal”. En un futuro cercano, con el advenimiento de nuevos implantes, será posible alargar las extremidades mediante cirugías simples, con una recuperación rápida y con un dolor mínimo para el paciente, lo que quizá permita cambiar nuestra postura frente a la talla que pueda alcanzarse.
Finalmente, es importante recalcar que las DE son enfermedades raras, que requieren de una alta sospecha clínica. En todo paciente con talla baja se debe ser acuciosos en la evaluación clínica de la desproporción, muchas veces sutil y un estudio radiológico, en manos expertas es clave en el diagnóstico. Si logramos un diagnostico especifico, luego de estudio de genes asociados, podremos ofrecer un manejo temprano, adecuado e integral.
No siempre se logrará una talla final normal, pero si se procurará mejorar la calidad de vida de nuestros pacientes. Se debe destacar que todo esto necesita de un correcto asesoramiento genético al paciente y su familia.
Referencias no citadas