



En la actualidad se conocen 8.000 enfermedades genéticas monogénicas. La mayoría de ellas son heterogéneas, por lo que el diagnóstico molecular por técnicas convencionales de secuenciación suele ser largo y costoso debido al gran número de genes implicados. El tiempo estimado para el diagnóstico molecular se encuentra entre 1 y 10 años, y este retraso impide que los pacientes reciban medidas terapéuticas y de rehabilitación específicas, que sus familiares entren en programas preventivos y que reciban asesoramiento genético.
La secuenciación masiva está cambiando el modelo de diagnóstico molecular de los afectos, sin embargo, los médicos y profesionales de la salud se enfrentan al dilema de la selección del método más eficiente, con el menor coste sanitario y con la mayor precisión de sus resultados. El objetivo de este trabajo es revisar la tecnología de secuenciación masiva y definir las ventajas y los problemas en su utilización.
Currently 8000 monogenic genetic diseases are known. Most of them are heterogeneous, so their molecular diagnosis by conventional sequencing techniques is labour intensive and time consuming due to the large number of genes involved. The estimated time is between 1 and 10 years for molecular diagnosis and this delay prevents patients from receiving therapy and rehabilitation measures, and their families from entering prevention programs and being given genetic counselling.
Next generation sequencing (NGS) is changing the model of molecular diagnosis of patients; however, doctors and health professionals are faced with the dilemma of choosing the most efficient method, with lower health care costs and the most accurate results. The aim of this paper is to review the NGS technology and define the advantages and problems in the use of this technology.
Según el catálogo OMIM (Online Mendelin Inheritance in Man), en la actualidad se conocen alrededor de 8.000 enfermedades genéticas relacionadas con todas las especialidades médicas, la mayoría de ellas de tipo monogénico. Sin embargo, su base genética se conoce en 4.423 entidades patológicas, lo que representa que en el 50% de enfermedades no se ha demostrado la relación genotipo-fenotipo1. Por otro lado, la Organización Mundial de la Salud (OMS), estima que estas 8.000 enfermedades afectan al 7% de la población mundial2. Según el informe de la Organización Europea de Enfermedades Raras (Eurordis), el 80% de las enfermedades raras son de origen genético3.
Las enfermedades monogénicas se heredan de forma mendeliana, siguiendo diversos patrones hereditarios conocidos: autosómico dominante, autosómico recesivo, ligados al X, ligados al cromosoma Y, disomía uniparental y trastornos de imprinting1. La mayoría están causadas por variantes patogénicas localizadas preferentemente en las regiones codificantes e intrónicas adyacentes de los genes, produciendo alteraciones de la proteína, modificando su función y provocando el fenotipo patológico. El diagnóstico genético se basa en la localización de dichas variantes en los genes asociados a las enfermedades.
Debido a la gran heterogeneidad genética que presentan las enfermedades monogénicas, el tiempo estimado para llegar al diagnóstico molecular se encuentra entre 1 y 10 años. Este retraso impide que los pacientes reciban medidas terapéuticas y de rehabilitación específica para su enfermedad, que sus familiares puedan entrar en programas preventivos dirigidos a la detección temprana de la enfermedad mediante estudios genéticos y que reciban asesoramiento genético sobre bases reales de la patología en estudio.
La estrategia actual de diagnóstico de enfermedades heterogéneas consiste en iniciar el estudio del gen más frecuentemente mutado asociado a la enfermedad mediante secuenciación Sanger. Si se detecta la variante patogénica, se confirma el diagnóstico molecular. Si no es así, se continúa con la secuenciación de los genes más frecuentemente mutados hasta detectar o no el gen y la mutación causante de la enfermedad.
La secuenciación masiva ha venido a solventar estos problemas dado que permite el análisis de todos los genes responsables de una enfermedad al mismo tiempo. Ello permite economizar tiempo y dinero, y establecer la etiología genética en la gran mayoría de las ocasiones.
El objetivo de este trabajo es conocer las tecnologías de secuenciación masiva aplicadas al diagnóstico y ayudar al médico a escoger el método molecular más rápido y de menor coste según la enfermedad del paciente.
1. TÉCNICAS ACTUALES DE ESTUDIOEn 1977, Fred Sanger y sus colaboradores publicaron dos artículos en los que describieron el método de secuenciación del ADN que ha transformado la biología de nuestros días y sigue siendo la tecnología de referencia. Se basa en la incorporación de didesoxinucleótidos trifosfato (ddNTPs) como terminadores de la cadena de ADN4,5.
Para llevar a cabo este proceso, se requiere ADN de cadena sencilla, un cebador de ADN, ADN polimerasa y ddNTPs con nucleótidos marcados con fluorescencia. Estos ddNTPs terminan la elongación de la cadena al carecer del grupo 3’-OH, produciendo varios fragmentos de ADN de longitud variable6.
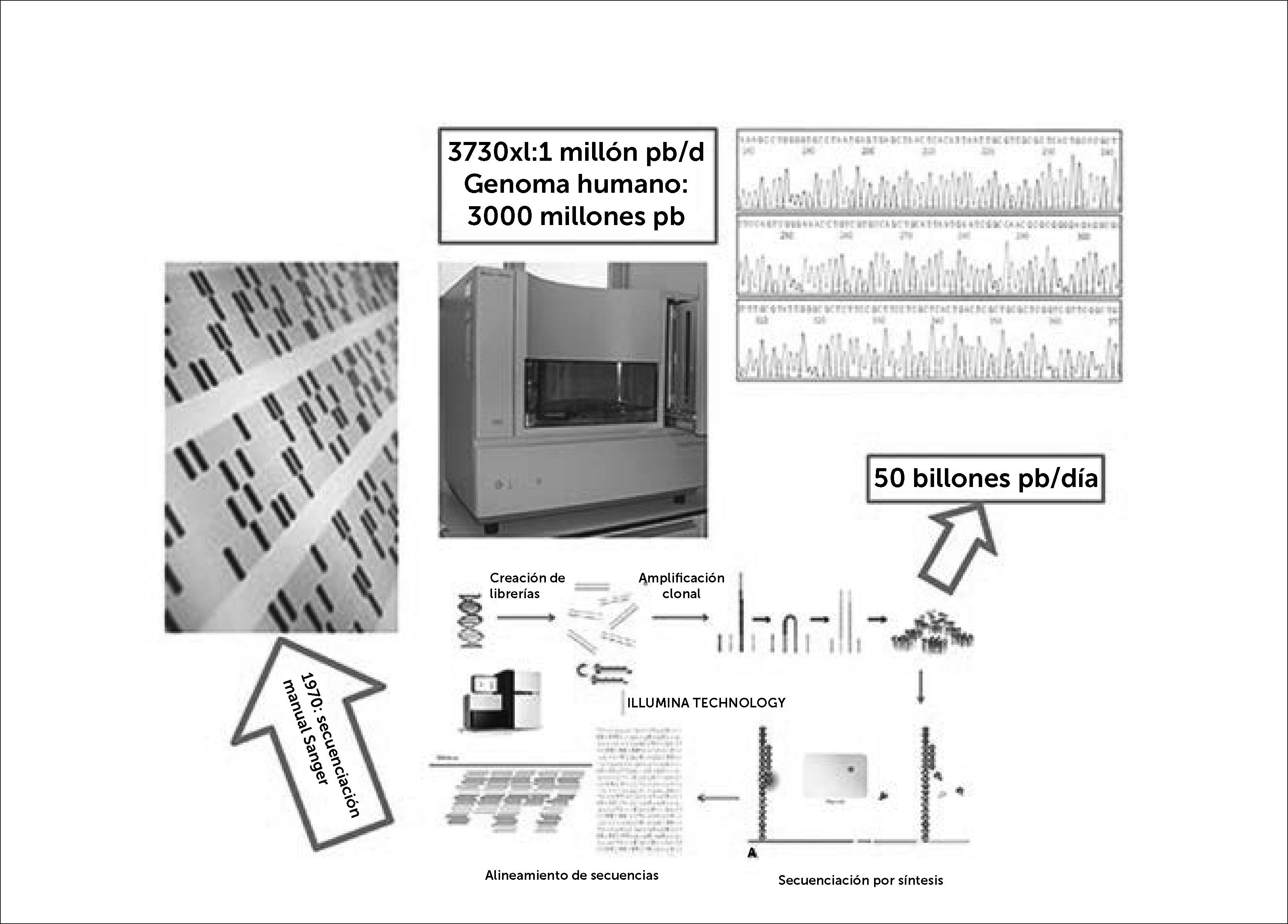
Desde su publicación, la secuenciación Sanger ha evolucionado de forma asombrosa permitiendo alcanzar hitos tan relevantes para la Genética Humana como la secuenciación del primer genoma humano en el año 20017, o la caracterización del primer haplotipo humano por el consorcio HapMap8. Sin embargo, tanto la inversión económica como el tiempo dedicado a estas tareas obligaron a desarrollar nuevas tecnologías de secuenciación más eficientes y baratas. De esta forma, surgieron las primeras plataformas de secuenciación masiva, abriendo una nueva era en las tecnologías de secuenciación y planteando un nuevo paradigma que se acaba de alcanzar al conseguir la secuenciación de un genoma por $1.000 9,10. Figura 1.

Evolución de la secuenciación Sanger manual a la secuenciación masiva
Desde la secuenciación manual de Sanger se evolucionó al Analizador de ADN 3730xl con capacidad de secuenciación de 1x106 pares de bases por día. En 45 años, los secuenciadores masivos pueden producir 50 billones de pares de bases por día.
La nueva generación de plataformas de secuenciación masiva o ‘Next-Generation Sequencing’ (NGS), inician su actividad comercial en el año 2005 generando una auténtica revolución en la investigación biológica. Si bien es cierto que la secuenciación tradicional mediante la tecnología Sanger ha dominado el campo de la investigación y el diagnóstico molecular durante al menos dos décadas, la vertiginosa evolución de estos nuevos secuenciadores, tanto en precisión como en rendimiento, así como el abaratamiento del coste por base han permitido la rápida expansión de su uso en la comunidad científica ofreciendo nuevas alternativas para la secuenciación de genomas completos 11–13, resecuenciación dirigida de zonas concretas del genoma 14–17, secuenciación de transcriptomas completo (RNA-Seq) 18, identificación de microRNAs 19, estudios de interacción proteína-DNA (ChIP-seq) 20 o estudios de metilación entre otros 21.
Durante la última década, se han realizado enormes progresos en términos de velocidad, longitud de lectura y rendimiento, por lo que se ha desarrollado un gran número de nuevas aplicaciones de NGS en ciencias básicas, así como en las áreas de investigación traslacional como los diagnósticos clínicos, agrogenómica y ciencias forenses 10.
A diferencia de la secuenciación Sanger, las plataformas de NGS permiten la obtención de miles o millones de fragmentos de ADN en un único proceso, haciendo posible el desarrollo de proyectos de secuenciación en corto plazo. A pesar de las diferencias en su química, todas estas plataformas de secuenciación masiva comparten las siguientes características:
- •
El ADN se fragmenta al azar y se unen adaptadores específicos a ambos lados de cada molécula directamente sin necesidad de clonar.
- •
La amplificación de la librería se produce mediante el anclaje del fragmento de ADN, a través de sus adaptadores, a una superficie sólida como son las microesferas o directamente a la placa de secuenciación.
- •
La secuenciación y detección de las bases ocurren al mismo tiempo en todas las moléculas de ADN (secuenciación masiva y paralela).
- •
Las lecturas generadas son cortas. El ruido producido por estas tecnologías en relación con la señal que generan limita la obtención de lecturas de mayor longitud.
- •
Las plataformas NGS permiten realizar secuenciación de tipo “paired-end” mediante la cual es posible leer los extremos del mismo fragmento de ADN. Esta estrategia de secuenciación facilita no solo el posicionamiento de aquellas lecturas que pueden mapear en múltiples sitios sino que también posibilita la identificación de variantes estructurales.
A pesar de todas estas particularidades comunes, cada plataforma de secuenciación masiva se basa en principios químicos distintos que generan diferencias cualitativas y cuantitativas.
2.1. Secuenciador 454-RocheFue la primera empresa en lanzar al mercado una plataforma de secuenciación masiva, cuya química está basada en la pirosecuenciación. Esta tecnología consiste en la detección de señales luminosas generadas a partir de grupos pirofosfato liberados tras la polimerización de un nuevo nucleótido complementario a una hebra de ADN molde. En sus inicios, este primer secuenciador despertó muchas dudas en la comunidad científica pese a presentar grandes ventajas respecto a la secuenciación Sanger, tales como el gran abaratamiento del coste de secuenciación por base (1/6 de lo que suponía en aquel entonces la secuenciación Sanger) y el gran aumento en la cantidad de información generada, equivalente a 50 secuenciadores Sanger. Más tarde, estos secuenciadores han evolucionado muy deprisa y actualmente la cantidad de información que generan es muy superior a su primera versión llegando a alcanzar 2Gb y una longitud de lectura de 700 a 1000 nucleótidos dependiendo de la versión 22.
2.2. Secuenciador SOLIDEl secuenciador SOLiD emplea una tecnología de ligación de oligonucleótidos marcados y es capaz de interrogar dos bases al mismo tiempo, de manera que, tras varias rondas de ligación y detección de fluoróforos, cada nucleótido es leído dos veces dando lugar a un nuevo tipo de codificación, que se denomina “double-encode” o “código de colores”, en el que cada color identifica dos bases consecutivas. Este proceso no se realiza con el primero y el último nucleótido. La longitud de las lecturas en la plataforma SOLiD alcanza los 75 nucleótidos y puede llegar a generar hasta 200 Gb de datos 23.
2.3. Secuenciadores de IlluminaLa plataforma Illumina se basa en la incorporación de nucleótidos marcados con terminadores reversibles de manera que en cada ciclo de ligación solamente uno de los cuatro nucleótidos posibles se une de forma complementaria al ADN molde emitiendo una señal luminosa que es captada por un sistema óptico altamente sensible. Posteriormente, el terminador se elimina para permitir la incorporación del siguiente nucleótido en ciclos sucesivos de secuenciación. La química empleada por Illumina permite generar lecturas de hasta 150nt llegando a producir hasta 1500 Gb en datos 24.
Por otro lado, la dificultad de reunir un número mínimo de muestras que justifiquen económicamente la puesta en marcha de un proceso completo unido al alto coste de adquisición de estos equipos impulsaron el desarrollo de una nueva serie de secuenciadores más económicos, capaces de generar una menor cantidad de datos con la misma precisión que las plataformas grandes de secuenciación masiva. En este contexto, se han desarrollado las siguientes plataformas: Secuenciador GS Junior de Roche, Secuenciador MiSeq, de Illumina, Secuenciador Ion Torrent de Applied. Estas plataformas se han extendido rápidamente dotando a pequeños laboratorios de la tecnología de secuenciación más avanzada.
2.4. Desarrollo de nuevas plataformas de tercera generaciónLas nuevas plataformas que se encuentran en desarrollo, conocidos como secuenciadores de tercera generación, permiten la secuenciación de una única molécula de ADN (single-molecule sequencing) evitando la amplificación de los fragmentos de ADN mediante PCR. De esta forma, se evitarían las desviaciones generadas durante el proceso de amplificación y, al mismo tiempo, se reduciría el tiempo de trabajo y el precio global de secuenciación. Algunas de estas plataformas son:
- -
Secuenciador SMRT de la empresa Pacific Biosciences, disponible comercialmente desde abril de 2011, basado en el uso de chips (tecnología Single Molecule, Real Time o SMRT) que contienen miles de pocillos en cuyo fondo se encuentra anclada una única proteína polimerasa que permite llevar a cabo la incorporación de nucleótidos marcados en tiempo real (750nt/sec) 25.
- -
Oxford Nanopores capaz de detectar nucleótidos individuales a su paso a través de un nanoporo 26.
Esta última generación de secuenciadores promete nuevas alternativas aún más baratas y con la posibilidad de resolver algunos problemas asociados a los secuenciadores actuales, tales como el estudio de expansiones, detección de variantes de número de copia o trastornos de metilación, así como la disminución de tiempo total de secuenciación. Sin embargo, continúan algunos desafíos significativos de NGS referidos al almacenamiento y procesamiento de datos.
3. DESDE LA INVESTIGACIÓN CIENTÍFICA HACIA EL DIAGNÓSTICO CLÍNICO MEDIANTE LA SECUENCIACIÓN MASIVATras la secuenciación del genoma humano los avances en la investigación y el desarrollo tecnológico han producido un profundo impacto en el progreso científico. El éxito del proyecto original dio lugar al lanzamiento de muchos otros esfuerzos internacionales cuyo objetivo común ha sido el profundizar en el conocimiento de la variabilidad genética entre individuos de la especie humana.
Uno de los proyectos más importantes de la última década ha sido el proyecto de HapMap iniciado en 2002 27, mediante un consorcio entre Japón, Reino Unido, Canadá, China, Nigeria y Estados Unidos. El objetivo de este proyecto fue identificar y catalogar las similitudes y diferencias de diferentes individuos de origen africano, asiático y de ascendencia europea, mediante comparación de sus secuencias genéticas. La identificación de regiones cromosómicas donde se comparten variantes genéticas permitiría encontrar genes implicados en enfermedades y variantes asociadas a respuesta a los diferentes fármacos. El proyecto de HapMap finalizó con la identificación de más de 8 millones de variantes comunes a lo largo del genoma, la mayoría de ellas localizadas mediante secuenciación Sanger 28, 29 y ha permitido la comparación de estas variantes con otros proyectos como 1000 genomas 30.
La llegada de las nuevas tecnologías de secuenciación ha aumentado generosamente la cantidad de datos y la velocidad de producción de los mismos. El proyecto “1000 genomes” fue pionero en emplear la secuenciación masiva de miles de individuos mediante tecnología NGS 31. Desde su inicio en 2007, este proyecto ha logrado determinar la localización y frecuencias alélicas de más de 15 millones de Single Nucleotide Variants (SNVs), un millón de inserciones y deleciones y 20.000 variantes estructurales, la mayoría de ellas no descritas previamente. Los resultados de este consorcio estiman que el 95% de la variabilidad de un individuo se encuentra presente en su base de datos y que cada persona lleva en su genoma entre 250-300 variantes de pérdida de función en genes anotados y de las cuales, entre 50 y 100 variantes se han asociado previamente a enfermedades hereditarias 32.
Durante los últimos años han surgido otras iniciativas internacionales que hacen uso de las nuevas plataformas de secuenciación como el “International Cancer Genome Consortium” (ICGC) 33, el proyecto “Cancer Genome Atlas” (TCGA) o el proyecto de ENCODE 34. Este último proyecto ha sido capaz de dar una imagen muy detallada de todos los transcritos primarios y maduros, así como la localización de las principales modificaciones de histonas, sitios de inicio y unión de factores de transcripción, sitios hipersensibles a DNAsa, descripción de más de 20.000 seudogenes, modificando el concepto de gen como la región genómica que codifica un conjunto de transcritos alternativos solapantes.
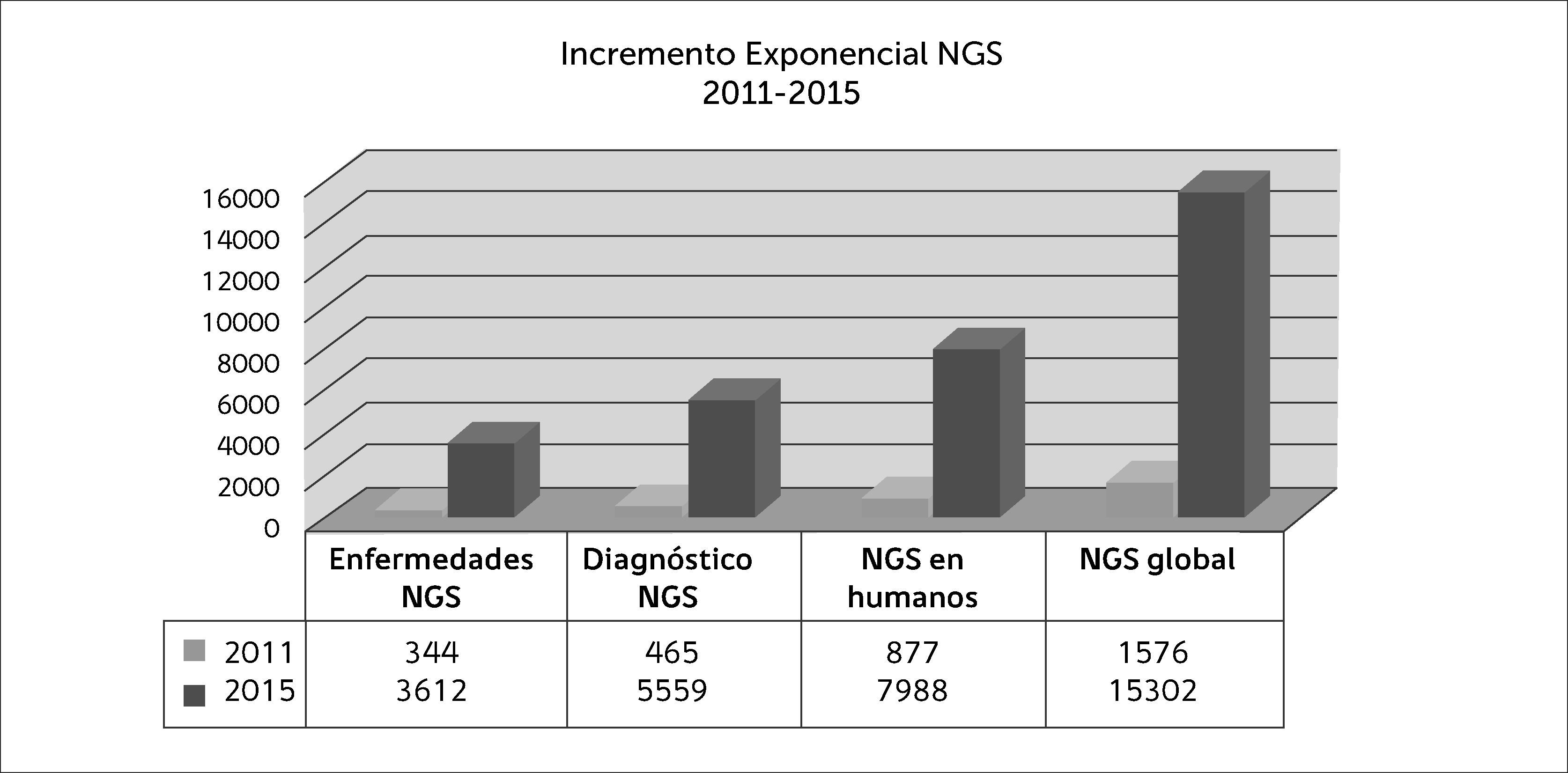
La gran cantidad de información generada por estos y otros consorcios de investigación ha sido depositada de forma paulatina en bases de datos públicas como dbSNP, que han visto cómo el número de variantes reportadas durante los últimos años se incrementaba de forma exponencial con la llegada de las nuevas tecnologías de secuenciación 35, así como la descripción de enfermedades monogénicas 36, Figura 2.

Secuenciación masiva en el diagnóstico de enfermedades monogénicas
El incremento exponencial de publicaciones entre 2011 y 2015 revela la ventaja del diagnóstico de enfermedades genéticas a través de la secuenciación masiva, así como la ampliación del conocimiento científico.
Fuente PubMed. Acceso 2 de mayo 2015.
El gran impacto provocado por la disponibilidad de esta valiosa información en bases de datos públicas así como la expansión del uso de estas plataformas, gracias a la caída en el precio de secuenciación por base, se ve claramente reflejado en el incremento exponencial del número de publicaciones de diversa índole basadas en el uso de NGS durante los últimos años.
El estudio y la integración de los datos disponibles provenientes de las tecnologías ómicas en diferentes bases de datos de dominio público han servido para la identificación de patrones genéticos comunes implicados en el desarrollo de enfermedades frecuentes en la población así como en la respuesta diferencial a fármacos o a determinados factores ambientales 37. La disponibilidad de la información generada por la comunidad científica de una forma organizada y estandarizada, es y será de vital importancia para poder realizar un diagnóstico más preciso, establecer un pronóstico precoz o inclusive, un tratamiento personalizado, si bien antes será necesaria la secuenciación de un mayor número de individuos que abarquen un abanico poblacional más amplio y el desarrollo de nuevos métodos de análisis que permitan explotar la información obtenida de forma exitosa.
4. EL DIAGNÓSTICO GENÉTICO MEDIANTE NGS: RESECUENCIACIÓN DIRIGIDA.VENTAJAS E INCONVENIENTESActualmente, el diagnóstico molecular en enfermedades monogénicas de origen genético conocido se realiza mediante la secuenciación bidireccional de exones y zonas de splicing mediante el método Sanger. Sin embargo, este procedimiento, ampliamente aceptado tanto por clínicos como por investigadores en genética, implica una serie de limitaciones en relación coste-eficiencia cuando se aplica a enfermedades con heterogeneidad genética donde no existe un único gen candidato sino un grupo de genes potencialmente candidatos 38. La posibilidad de diseñar sistemas de captura “a medida” dirigidos a la selección de regiones específicas del genoma ha abierto una nueva perspectiva en el diagnóstico genético en este tipo de enfermedades. La mejora continua de la química de secuenciación así como de los protocolos de trabajo y la automatización de los mismos han permitido perfeccionar la precisión y el rendimiento de las plataformas de secuenciación masiva dando lugar a un aumento en la cantidad de datos generados y a un descenso vertiginoso del coste de secuenciación por base. Sin embargo, pese a que actualmente el precio de secuenciar un genoma humano completo sea una ínfima parte de lo que costó la obtención del primer borrador en 2001, la secuenciación de rutina de genomas completos todavía sigue siendo económicamente inabordable para la mayoría de las instituciones.
Uno de los desarrollos que mayor impacto ha tenido en el estudio de genomas a gran escala mediante secuenciación masiva, es la posibilidad de capturar zonas concretas del genoma, técnica conocida como resecuenciación dirigida, secuenciación masiva paralela o TargetSeq. Durante los últimos años se han desarrollado distintas técnicas de captura de ADN basadas en la hibridación de sondas o en sistemas de PCR multiplex 39,40 que permiten abarcar desde pocas kilobases hasta la captura del exoma completo, y que, combinados con la secuenciación masiva hacen posible realizar un screening mutacional en cientos de genes a la vez, proporcionando una cantidad de información muy extensa sobre la genética de un individuo y acortando el tiempo de diagnóstico molecular de las enfermedades genéticas heterogéneas.
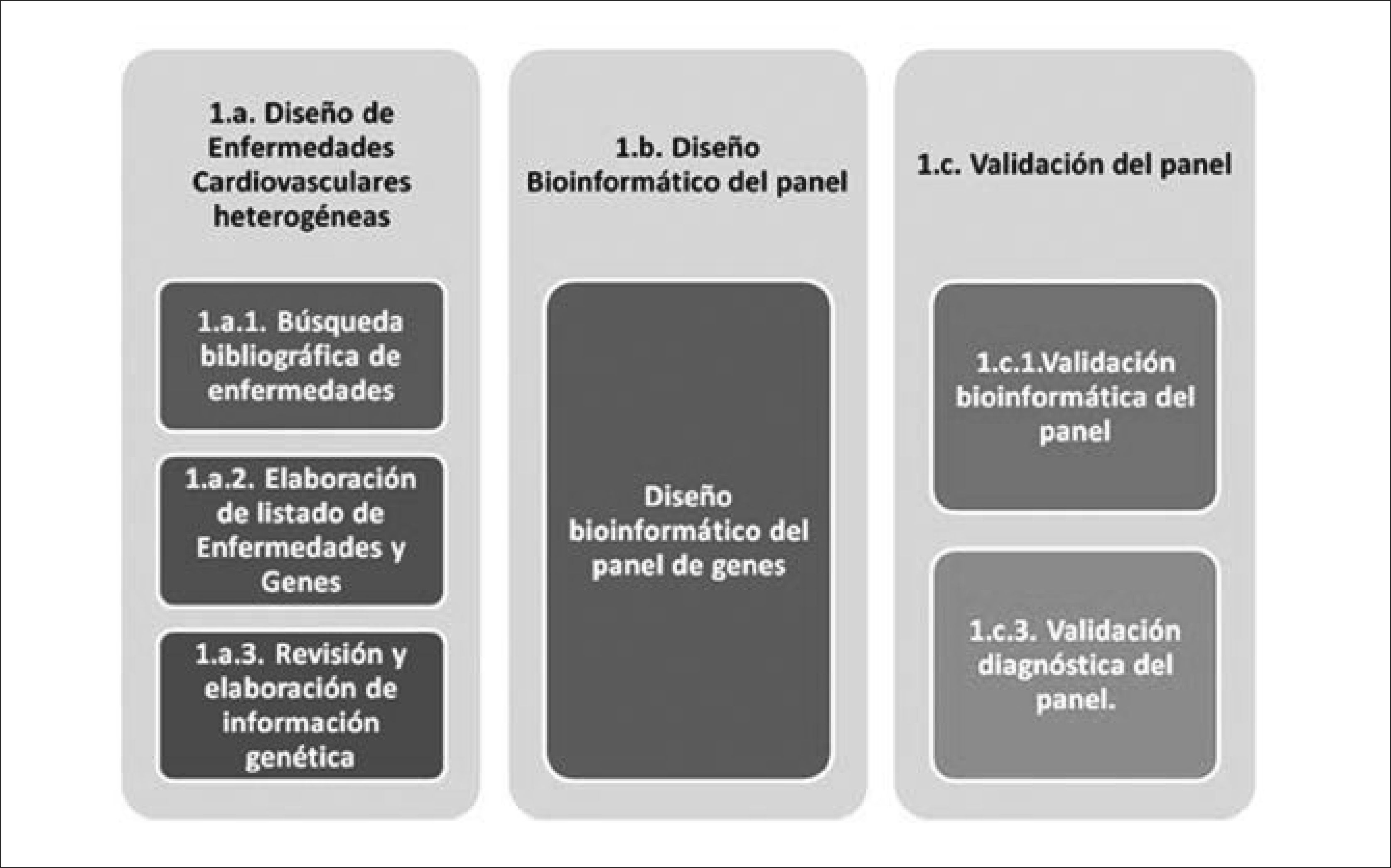
La aplicación de paneles de secuenciación masiva al diagnóstico genético de enfermedades genéticas heterogéneas requiere de un proceso de diseño biomédico de los genes asociados a la patología, del diseño bioinformático del panel y de la validación bioinformática y diagnóstica del panel que permita evaluar los parámetros de calidad del panel como la reproducibilidad, cobertura media, sensibilidad, especificidad, detección de deleciones e indels, confirmación de variantes previamente estudiadas por secuenciación Sanger, previa a su aplicación en el diagnóstico de enfermedades genéticas 41,42. En la figura 3 se encuentra el modelo de diseño y validación del panel para Enfermedades Cardiovasculares.

Modelo de diseño y validación del panel de enfermedades cardiovasculares heterogéneas
La fase 1.a. corresponde al diseño de un panel de genes aplicados a enfermedades cardiovasculares. Incluye la búsqueda de información clínica y genética. En la fase 1.b. se encuentra el diseño bioinformático del panel para su fabricación. La fase 1.c corresponde a la evaluación bioinformática del diseño y a la validación diagnóstica con muestras clínicas conocidas para establecer su sensibilidad y especificidad.
La resecuenciación dirigida de un pequeño número de genes implica una reducción significativa en la cantidad de lecturas necesarias para la identificación de las variantes presentes con el consecuente la disminución en el coste por muestra. La llegada de los mini-secuenciadores con tecnología NGS así como la disminución de los costes de secuenciación por base, forman la combinación ideal para la adopción de esta tecnología como método de preferencia en el diagnóstico genético de rutina en el presente y futuro cercano 43.
La descripción de enfermedades genéticas heterogéneas se encuentran en todas las especialidades médicas y están asociadas a mutaciones en múltiples genes, lo que ha puesto sobre la mesa la dificultad tecnológica de estudiar de forma eficiente, todos los genes implicados en una patología.
La mayoría de estudios moleculares se realizan de acuerdo a la frecuencia descrita de mutaciones en determinados genes, produciendo un registro incrementado de mutaciones sobre los genes estudiados y un sub registro de mutaciones en genes no estudiados, provocando una disminución de frecuencias de asociación de estas enfermedades a dichos genes.
Además, debido al tamaño y complejidad de ciertos genes, no ha sido posible su estudio por secuenciación Sanger, lo que ha determinado la imposibilidad de conocer su verdadera implicación en las enfermedades heterogéneas, tal es el caso del gen TTN con 313 exones, cuyas mutaciones pueden producir Miocardiopatía dilatada, Displasia arritmogénica ventricular, Miocardiopatía hipertrófica y diversos tipos de miopatías y distrofias musculares con diversos grados de severidad y modelo hereditario.
Uno de los mayores avances en estos últimos años, ha sido definir la base genética de enfermedades cardiovasculares, particularmente asociadas a muerte súbita. Hoy se conocen más de 40 enfermedades cardiovasculares en las cuales se han identificado una causa genética, entre ellas, miocardiopatías de diferentes tipos, diversos trastornos del ritmo cardíaco incluyendo varias canalopatías y un grupo de trastornos asociados a aneurismas de aorta torácica, con la identificación de más de 200 genes para los que se han desarrollado paneles de estudio por resecuenciación dirigida.
Actualmente, este tipo de paneles se aplican en enfermedades neurogenéticas como Charcot-Marie-Tooth, ataxias, paraplejias, miastenia, miopatías, distrofias musculares. Igualmente se han desarrollado paneles para hipoacusias, cáncer familiar incluyendo genes de alta y moderada penetrancia, trastornos visuales, epilepsia, discapacidad intelectual, enfermedades mitocondriales, enfermedades metabólicas, incrementando la tasa de pacientes diagnosticados desde el punto de vista molecular.
NGS utiliza una combinación de nuevas estrategias en la preparación de muestras de ácidos nucleicos, nuevos métodos de secuenciación de ADN y nuevos enfoques para la alineación y el montaje del genoma, siendo posible un análisis genético de todos los genes descritos en pocas semanas, disminuyendo su tiempo de estudio que previamente era de meses o años, mejorando la efectividad y eficiencia en el diagnóstico, al identificar un alto número de variantes de ADN que pueden actuar como mutaciones patogénicas responsables de la enfermedad o como modificadores de la expresividad fenotípica 44. Estos avances tienen mayores implicaciones para la práctica clínica ya que se necesita integrar estos hallazgos genéticos en el manejo clínico de los pacientes como herramienta diagnóstica y, en el futuro, establecer el pronóstico y posibles tratamientos personalizados, sin dejar de lado la detección de familiares en riesgo que deben integrarse a programas preventivos de la enfermedad y el asesoramiento genético familiar sobre bases moleculares concretas.
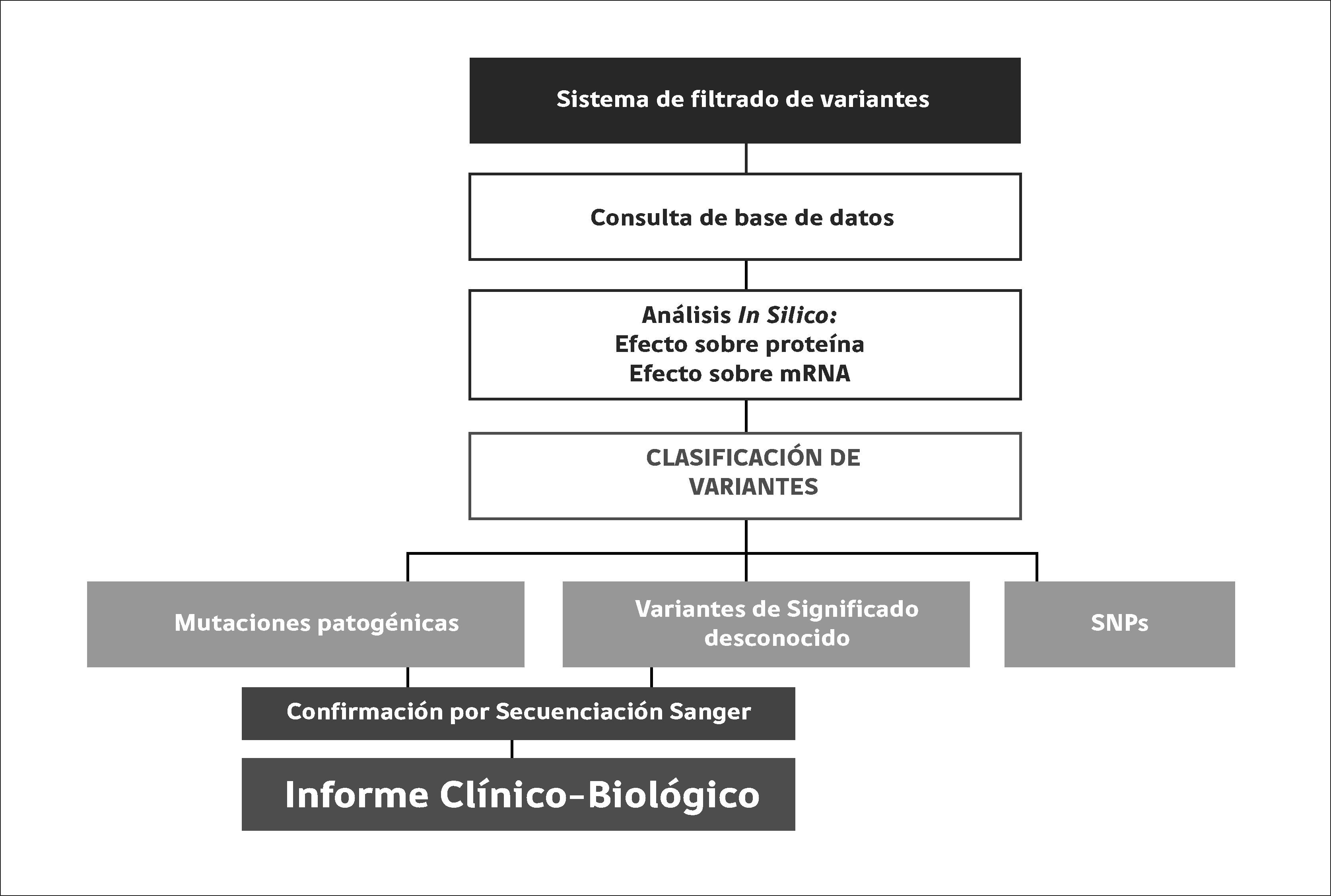
Sin embargo, es indispensable que el diagnóstico molecular aplicando paneles de resecuenciación dirigida sea validado previamente, estableciendo flujogramas de trabajo consensuados que lleven a la interpretación correcta de las variantes detectadas, a la integración a bases de datos de forma automática y a la revisión de estudios anteriores 45, Figura 4.

Flujograma de diagnóstico clínico-biológico en paneles de secuenciación masiva
La clasificación de variantes detectadas por secuenciación masiva incluye la aplicación de algoritmos validados y actualización constante de información científica y guías consensuadas, que permitan definir las variantes patogénicas y probablemente patogénicas que requieren ser confirmadas previamente a la emisión del informe clínico biológico.
Aunque estos avances han aportado un nuevo conocimiento, es necesario implementar guías consensuadas para el manejo de la enorme cantidad de información genética generada para el análisis, la interpretación biológica y clínica y la información al paciente y su familia. Igualmente, son indispensables nuevos desarrollos y actualizaciones de bioinformática, estudios de cosegregación familiar, demostración de ausencia de la variante en la población normal y estudios funcionales que permitan determinar el significado biológico y la patogenicidad de las variantes identificadas, así como su integración en bases de datos que permitan la correlación genotipo-fenotipo.
Además, es necesario definir los aspectos bioéticos respecto del derecho a conocer nueva información sobre genes que no se encuentran en el panel de estudio, pero que a la vez, puede ayudar en un nuevo diagnóstico de la enfermedad, ya sea por fenotipos solapantes o por ausencia de signos a la fecha de estudio, por lo que es indispensable el abordaje multidisciplinario para establecer la correlación genotipo-fenotipo del afecto así como el manejo de familias con enfermedades genéticas heterogéneas 46.
La gran trascendencia de los resultados en el ámbito familiar permite asesorar con alta precisión a aquellos familiares en riesgo, estableciendo medidas de prevención dirigidas, así como disminuir la angustia en aquellos familiares con un riesgo bajo. La detección de los factores de riesgo también va a influir en el asesoramiento reproductivo y en las recomendaciones de diagnóstico prenatal, diagnóstico preimplantación u otras opciones reproductivas.
Next Generation BioinformaticsLa transición desde la secuenciación tradicional automatizada de Sanger a plataformas con una mayor capacidad de producción de datos ha forzado el desarrollo de nuevos algoritmos bioinformáticos y métodos de análisis necesarios para la correcta manipulación e interpretación de una nueva generación de datos. Los continuos aumentos en la capacidad de generación de datos que se duplica aproximadamente cada cinco meses, ha sobrepasado con creces los recursos bioinformáticos disponibles hasta la fecha, suceso que algunos autores han denominado como “Next-Generation gap” 47.
Sin lugar a dudas, el desarrollo de las plataformas de Next- Generation Sequencing ha desencadenado el desarrollo de “Next-Generation Bioinformatics” que se enfrenta no solo a un gran reto a nivel informático sino también biológico ya que por primera vez es posible disponer de la información total de un individuo. El desarrollo de herramientas bioinformáticas es fundamental para la aplicación con éxito de la secuenciación masiva al diagnóstico genético.
Del mismo modo que la generación de datos es un proceso protocolizado y moderadamente estable para cada plataforma, el análisis de los datos no sigue un modelo “gold-standard”, y la combinación de diferentes piezas de software así como su integración con diferentes bases de datos proporcionan resultados muy distintos. La correcta identificación y caracterización del conjunto de variantes presentes en una muestra es uno de los procesos clave y más sensible para la correcta aplicación de la secuenciación masiva en el diagnóstico genético no solo debido a la enorme cantidad de datos que han de manipularse al mismo tiempo sino también por la gran complejidad biológica que plantea 48. El desarrollo y establecimiento de protocolos de trabajo eficientes, precisos y validados que permitan garantizar unos estándares de calidad es una tarea larga y compleja, debido a la necesidad de un exhaustivo conocimiento del funcionamiento de las plataformas de NGS así como de la biología del sistema en estudio para avanzar en el conocimiento sobre la complejidad del genoma humano y progresar así hacia un diagnóstico más preciso, un pronóstico más precoz, y un tratamiento personalizado. Las primeras aplicaciones ya están dando resultados en muchos aspectos de nuestra vida cotidiana, que fundamentalmente van a afectar de forma directa en tres áreas: las políticas sanitarias, el diagnóstico médico y el tratamiento 49–51.
5. DESDE EL DIAGNÓSTICO GENÉTICO HACIA EL DIAGNÓSTICO GENÓMICO. INDICACIONES DE ESTUDIOLa secuenciación masiva está cambiando el modelo de diagnóstico molecular de los pacientes afectos de patología genética, de tal manera que los médicos y profesionales de la salud se enfrentan al dilema de la selección del mejor método de diagnóstico para la patología genética de sus pacientes y de la comunicación de sus resultados. Por lo tanto, es indispensable comprender las ventajas y los problemas de las diferentes tecnologías, la interpretación de sus resultados, el significado clínico de las variantes detectadas, los problemas éticos que se derivan.
Debido al gran número de pruebas moleculares disponibles, tanto genéticas (secuenciación Sanger, estudio de expansiones por análisis de fragmentos, pruebas de metilación, estudios de disomía uniparental, amplificación múltiple de sondas dependientes de ligación entre otras) así como genómicas (array de hibridación genómica comparada con o sin polimorfismos de un solo nucleótido, paneles de secuenciación masiva, exoma, genoma) se han establecido modelos de estudio en las enfermedades mendelianas 52,53.1. Estudios de un solo gen: se recomienda este tipo de estudios en las siguientes condiciones:
- -
Cuando la enfermedad se produce por mutaciones específicas producidas en un solo gen, como es Acondroplasia producida en el 99% de casos por las mutaciones c.1138G>A (p.Gly380Arg) o c.1138G>C (p.Gly380Arg) del gen FGFR3.
- -
Cuando la enfermedad tiene heterogeneidad mínima, como es la fibrosis quística producida por el gen CFTR.
- -
Cuando la metodología molecular a utilizar es diferente de secuenciación Sanger, por ejemplo estudio de expansiones en Ataxias espinocerebelosas o síndrome de X Frágil, MLPA en Distrofia muscular de Duchenne/Becker o estudios de metilación en síndromes de Prader Willi o Angelman.
2. Paneles de secuenciación masiva paralela: se utilizan preferentemente en las siguientes condiciones:
- -
Enfermedad con heterogeneidad genética: en este grupo se incluyen la mayor parte de enfermedades mendelianas en las diferentes especialidades médicas: enfermedades cardiogenéticas como miocardiopatías trastornos del ritmo cardiaco, aneurisma de aorta; cáncer familiar, enfermedades neurogenéticas como ataxias, Charcot Marie Tooth, distrofias musculares, paraplejia espástica, distonías, Parkinson; discapacidad intelectual, epilepsias; trastornos mitocondriales de regulación nuclear, trastornos metabólicos, hipoacusias, trastornos visuales, displasias esqueléticas, etc.
- -
Se incluyen trastornos solapantes que se encuentran en el diagnóstico diferencial de la patología genética, tal es el caso de miocardiopatías.
- -
Trastornos que comparten manifestaciones clínicas, pero pueden tener diferencias en la presentación global de la enfermedad, como es el caso de epilepsias.
- -
Enfermedades producidas por alteración de una vía común, tal es el caso de RASopatías.
3. Exoma: se utiliza en patología con extrema heterogeneidad, en pacientes con dos o más fenotipos no relacionados o ausencia de características clínicas claves al momento de la prueba. Sin embargo, todavía deben desarrollarse estrategias de análisis más eficiente, las diferencias de cobertura que tiene el exoma y la interpretación de resultados.
ConclusiónLa secuenciación masiva aporta nuevo conocimiento genético, permite realizar el diagnóstico molecular de un número mayor de pacientes, explicando la variabilidad fenotípica, ayuda a establecer medidas preventivas en familiares en riesgo, acceder a asesoramiento genético familiar y a disminución de costes sanitarios en familiares que se encuentran en el riesgo poblacional.
6. PERSPECTIVAS FUTURASEl avance vertiginoso de las tecnologías de secuenciación masiva se dirige hacia el diagnóstico con especificidad cercana al 100%, al mejorar los algoritmos bioinformáticos de estudio en donde no sea necesario la confirmación de variantes patogénicas por medio de secuenciación Sanger 38,54, hacia el estudio de cualquier trastorno molecular en un solo proceso, tal es el caso de expansiones, deleciones o duplicaciones.
Se espera que el estudio del Genoma aporte la detección de variantes patogénicas en regiones no estudiadas por los métodos actuales así como la detección de variantes en el número de copias genómicas y se comprenda la influencia de las regiones reguladoras, transformándose en la única prueba genética para el diagnóstico y seguimiento de pacientes.
También el desarrollo de bases de datos de población normal y de enfermedades, sus mecanismos de producción, los estudios funcionales, bases de datos de elementos reguladores, desarrollo del transcriptoma y metiloma darán su aporte al conocimiento de la medicina genómica y su aplicación en medicina personalizada desde el diagnóstico hacia el tratamiento, modificando el modelo de la práctica médica con nuevas oportunidades y retos.
No obstante, la investigación traslacional debe aclarar la interpretación de las variantes, el almacenamiento de los datos bioinformáticos, la insuficiencia de los procedimientos de consentimiento informado para las pruebas genéticas, las plantillas limitadas de genetistas clínicos y de laboratorio, abriendo nuevas oportunidades a todos los profesionales de la salud ya que nos encontramos frente al enorme potencial de esta nueva tecnología.
Los autores declaran no tener conflictos de interés, en relación a este artículo.