




Dismorfología se refiere al estudio de los pacientes con malformaciones congénitas. En este concepto se incluyen también pacientes con otras alteraciones morfológicas que lo hacen aparecer diferente. En este artículo revisamos los diferentes tipos de alteraciones que el médico debe reconocer, tanto cualitativas, como malformaciones, deformaciones, disrupciones y displasias, como cuantitativas enfatizando la importancia de diferenciar si éstas constituyen variación normal, racial o familiar, o son indicadores de una afección genética. Delinearemos la forma de estudiar al paciente y los problemas más frecuentes que dificultan establecer el diagnóstico. Si la malformación es aislada, de causa poligénica/multifactorial, es habitualmente el médico tratante quien establece el diagnóstico y otorga asesoramiento genético. En caso de anomalías múltiples, en que hay que determinar su etiología específica, lo recomendable es referir el paciente a un genetista clínico. Sólo así se podrá dar asesoramiento genético responsable ayudando al paciente a lograr su máximo potencial genético y a la familia a tener hijos normales.
Dysmorphology refers to the study of patients with congenital malformations. However, dysmorphology not only includes the study of birth defects but also the study of patients with other anomalies making him/her look different. In this article we review qualitative abnormalities, such us malformations, deformations, disruptions and displasias as well as quantitative variations which may represent normal, racial or familial, variation or be part of a genetic disorder. We review the study of the patient and frequent diagnostic problems. If the congenital anomaly is isolated, non-syndromic, of polygenic/multifactorial etiology, it is the responsibility of the primary physician to establish the diagnosis and provide genetic counseling. However, in cases of multiple anomalies when the physician should establish the specific etiology, the recommendation is to refer the patient to a clinical geneticist. The main goal of the evaluation is to determine the etiology of the abnormalities. Only then can the physician provide responsible genetic counseling, helping the patient to achieve his/her maximum genetic potential and allow the family to have normal children.
Dismorfología (dis=anormal, morfo=forma, estructura) fue el término acuñado por David Smith en los años sesenta para referirse en general al estudio de los pacientes con malformaciones congénitas. Sin embargo, este campo incluye también pacientes con alteraciones cuantitativas, asimetrías, etc. que hacen aparecer al paciente diferente. Debemos mencionar que el uso de la sigla en inglés FLK (por “funny-looking kid”) está proscrito y no debe usarse por la connotación peyorativa que tiene para las familias. El propósito de este artículo es familiarizar al médico general con la evaluación del paciente dismórfico. Revisaremos las alteraciones cualitativas, como malformaciones, deformaciones, disrupciones y displasias; enfatizaremos la importancia de detectar alteraciones cuantitativas determinando si éstas constituyen variaciones normales, raciales o familiares, o si son indicadores de una afección genética. En caso de anomalías múltiples, determinar si las alteraciones constituyen parte de un síndrome, una secuencia o de un defecto de campo monotópico o politópico. El concepto de asociación, inicialmente descrito como un concepto estadístico, está cambiando por entidades biológicas o sindromáticas al identificarse las alteraciones moleculares subyacentes. El objetivo principal de la evaluación es determinar la etiología de estas alteraciones. Sólo así se podrá dar asesoramiento genético responsable.
En la práctica clínica, el médico se puede ver enfrentado a tres tipos de afecciones genéticas:
Estructurales, que corresponden al campo de la dismorfología.
Funcionales, como son algunos errores congénitos del metabolismo, o la presencia de sordera, ceguera, u otra alteración funcional.
Un grupo mixto, de reconocimiento relativamente reciente, en que el mejor ejemplo es el síndrome de Smith-Lemli-Opitz. Este es un cuadro de malformaciones congénitas múltiples debido a un defecto en el metabolismo del colesterol. En esta actualización revisaremos el enfoque del médico enfrentado al paciente dismórfico que es ciertamente la situación más frecuente en la práctica clínica.
Está bien establecido que en cualquier pais, cuando la mortalidad infantil baja de veinte por mil, las anomalías congénitas constituyen la primera causa de mortalidad. Si bien generalmente se acepta una frecuencia de malformaciones congénitas de 3% (NCBDDD, 2014), si uno examina cuidadosamente a todos los recién nacidos, su frecuencia alcanza aproximadamente un 8%. Si se incluyen malformaciones detectadas postnatal o se incluyen anomalías menores, el porcentaje aumenta a más del 15%. Si bien los pacientes dismórficos frecuentemente presentan malformaciones severas que afectan múltiples órganos o sistemas y tienen un impacto importante en la morbilidad y mortalidad infantil, así como en la familia y sociedad, su evaluación diagnóstica es de máxima importancia para proporcionar consejo o asesoramiento genético y prevenir su recurrencia en la familia. Un diagnóstico correcto es la piedra angular. Cuando se habla de diagnóstico en genética clínica nos referimos al diagnóstico etiológico. No basta el diagnóstico fenotípico (como sería el diagnóstico de enanismo, parálisis cerebral, discapacidad intelectual o múltiples malformaciones congénitas) ni patogénico, como son las secuencias. De aquí que el autor, en 1994, sugirió el uso de un sistema internacional de diagnóstico usando a lo menos tres ejes:
i) El diagnóstico fenotípico
ii) El diagnóstico patogenético
iii) El diagnóstico etiológico. Si éste no está claro se puede incluir un cuarto eje con los diagnósticos diferenciales.
Para establecer el diagnóstico habitualmente hay que realizar una evaluación completa que incluye historia familiar detallada, historia prenatal, natal, perinatal y postnatal, y un examen físico detallado que debe ser objetivo, discriminativo y completo. Para esto recomendamos ir en orden secuencial. En el examen físico debemos determinar el tipo de anomalía, vale decir, si ésta o éstas son cualitativas, como lo son las malformaciones o disrupciones, deformaciones o displasias, o cuantitativas.
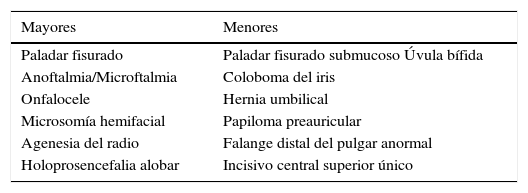
Malformaciones son aquellos defectos morfológicos de un órgano, parte de un órgano o de una zona mayor del cuerpo, resultante de un proceso de desarrollo intrínsecamente anormal. Las malformaciones pueden ser mayores o menores. Mayores son aquellas con importantes consecuencias médicas, quirúrgicas o cosméticas. Menores son aquellas sin dichas características aunque a veces tienen cierto impacto cosmético. En todo caso, las malformaciones menores tienen el mismo significado señalando una morfogénesis anormal. Las malformaciones menores son más frecuentes, pueden implicar defectos estructurales severos y frecuentemente constituyen una clave diagnóstica importante (Tabla 1).
EQUIVALENCIA ENTRE MALFORMACIONES MAYORES Y MENORES
| Mayores | Menores |
|---|---|
| Paladar fisurado | Paladar fisurado submucoso Úvula bífida |
| Anoftalmia/Microftalmia | Coloboma del iris |
| Onfalocele | Hernia umbilical |
| Microsomía hemifacial | Papiloma preauricular |
| Agenesia del radio | Falange distal del pulgar anormal |
| Holoprosencefalia alobar | Incisivo central superior único |
Las deformaciones son causadas por fuerzas mecánicas intrínsecas o extrínsecas, como es lo que ocurre en el pie bot y tienen mejor pronóstico dado que tienen una mejor respuesta al tratamiento.
Las disrupciones son a veces difíciles de distinguir de las malformaciones pues pueden producir un fenotipo similar; pero en este caso son defectos morfológicos de un órgano o parte de un órgano que resultan de la interferencia al proceso de desarrollo normal. Ejemplo clásico de disrupciones son las bandas amnióticas. Las disrupciones de origen vascular no son tan infrecuentes.
Las displasias son el proceso y la consecuencia de una formación u organización anormal de las células en los tejidos. Displasias como nevus o hemangiomas, son muy comunes.
En dismorfología, las alteraciones cuantitativas, también llamadas anomalías menores (existen diferentes clasificaciones y nomenclaturas de acuerdo a diferentes expertos) son muy comunes (14% a 15% de la población general) y pueden representar características familiares o raciales normales o ser manifestaciones de afecciones genéticas. De aquí la importancia de examinar o evaluar a padres, hermanos y otros familiares. Mientras más anomalías menores tenga el paciente, mayor es la probabilidad de que tenga alguna anomalía mayor y por lo tanto una afección genética. Hay que recordar que la mayoría de los rasgos faciales característicos de muchos síndromes genéticos, por ejemplo el síndrome de Down, son anomalías menores y no malformaciones. Un elemento diagnóstico importante en dismorfología está dado por las asimetrías, desproporción de segmentos corporales y crecimiento anormal. De aquí que un examen físico apropiado sea tan importante. En general, en casos de crecimiento global aumentado o disminuido es conveniente consultar a un endocrinólogo. Si hay hemihipotrofia debemos investigar la posibilidad de un mosaicismo; si hay hemihipertrofia hay que descartar la posibilidad de tumores.
Si el paciente presenta una anomalía aislada como labio y paladar hendido, defecto del cierre del tubo neural o una cardiopatía congénita aislada, el diagnóstico es simple y lo más frecuente es que sea una alteración poligénica/multifactorial con un bajo riesgo de recurrencia. Habitualmente estos pacientes son manejados por el pediatra general, médico de familia o especialista, pero si el paciente presenta múltiples anomalías congénitas debería ser referido a un centro de genética. El genetista le diagnosticará si las alteraciones corresponden a un síndrome, secuencia o simplemente a un defecto de campo monotópico o politópico.
En caso de las secuencias, hay que tratar de determinar la causa de la anomalía original que desencadenó la cascada de anomalías secundarias resultantes de la interferencia del proceso de desarrollo embrionario. Por ejemplo, en el caso de la secuencia de Pierre-Robin (micrognatia o mandíbula pequeña, paladar hendido y desplazamiento hacia abajo de la lengua), necesitamos determinar la causa inicial que produjo una hipoplasia mandibular antes de las nueve semanas de desarrollo intrauterino. Sabemos que aproximadamente un tercio de estos casos son debidos a mutaciones en uno de los genes codificadores de colágeno y corresponden a un síndrome de Stickler.
Si bien síndrome (sin = junto; drome = correr) significa manifestaciones, síntomas o signos que ocurren juntos y el término se usa frecuentemente en medicina, como cuando se habla de síndrome febril, en genética se usa el concepto cuando existe una causa o etiología determinada que explica la condición. El concepto de asociación, inicialmente descrito como un concepto estadístico más que biológico, está cambiando a medida que se reconoce la causa de estas blastopatías. Así por ejemplo, la asociación CHARGE es considerada ahora un síndrome debido a que fue posible determinar era producida por una mutación en el gen SEMA3E.
La etiología de las malformaciones puede ser genética y/o ambiental. Clásicamente como genéticas se consideraban las afecciones de causa mendeliana o monogénicas, cromosómicas y poligénicas/multifactoriales; y como no genéticas o ambientales a los teratógenos. Estos no sólo incluyen ciertas drogas, radiaciones, virus, alcoholismo, sino incluyen también condiciones maternas como la diabetes. Sin embargo, en los últimos 30 años, además de estas etiologías clásicas se han reconocido nuevas, no tradicionales, como los síndromes de genes contiguos, los debidos a mutaciones dinámicas (como el síndrome del X-Frágil), a mecanismo epigenético, herencia trialélica, mitocondrial, entre otras.
A pesar de los avances tecnológicos y los nuevos exámenes de laboratorio disponibles, incluyendo microrrays, secuencia exónica, y, en un futuro muy cercano, secuencia de todo el genoma, una buena evaluación clínica continúa siendo lo más importante en la evaluación diagnóstica. Ciertamente estos nuevos exámenes junto con confirmar la etiología, están comenzando a cambiar las descripciones clásicas de algunos síndromes.
El conocido pediatra-hematólogo Frank Oski, quien falleciera en 1996, describió cuatro formas de establecer el diagnóstico:
1) Reconocimiento de las características clínicas, ya sea por el fenómeno gestáltico o asociación de anomalías.
2) A través de una hipótesis diagnóstica.
3) El uso de algoritmos.4) Lo que él denominara “testing the universe” que se refiere a tratar de establecer un diagnóstico haciendo todo tipo de exámenes con la esperanza de que alguno de ellos revele el diagnóstico. Si bien una buena cantidad de síndromes dismórficos son fácil e inmediatamente diagnosticados por sus evidentes características fenotípicas, sea por el fenómeno gestáltico o asociación, la mayoría de los pacientes dismórficos requiere una detallada evaluación clínica siguiendo una hipótesis diagnóstica como el Dr. Oski señalara. En diferentes publicaciones de textos nacionales (ver referencias) así como en la revista Médico Interamericano, hemos enfatizado la importancia de una adecuada y completa historia familiar, prenatal, natal, perinatal y postnatal, así como de un detallado examen físico. En algunos de esos capítulos hemos incluido algunos acrónimos creados para ayudar a una evaluación más completa. El examen dismorfológico de la cara, manos y pies, incluyendo el estudio de los dermatoglifos, ha demostrado ser de la mayor importancia y rendimiento. El análisis de los dermatoglifos que consiste en el estudio de los surcos epidérmicos y los patrones que forman en los dedos, palmas, ortejos y planta de los pies, no sólo permite establecer algunos diagnósticos como el síndrome de Down, sino con frecuencia sugieren la existencia de una alteración cromosómica (Tabla 2), un posible mosaicismo, o la presencia de algún síndrome específico. Esta evaluación, siguiendo una hipótesis diagnóstica, es lo que se debe enseñar en las escuelas de medicina. En dismorfología sólo ocasionalmente usamos algoritmos clínicos, y siempre, siguiendo al Dr. Oski, desaconsejamos a los médicos el solicitar en forma indiscriminada exámenes con la esperanza de que alguno les dé el diagnóstico. Esta práctica inapropiada de establecer un diagnóstico es un factor importante que contribuye al excesivo costo de la medicina en estos días. Dependiendo de la sospecha diagnóstica específica se deberá elegir qué tipo de examen citogenético, molecular o metabólico se deberá solicitar para confirmar la sospecha diagnóstica. Obviamente, previo a solicitar exámenes genéticos, se deben efectuar consultas a los diferentes especialistas que estén involucrados, solicitar radiografías u otros exámenes específicos habituales, con la finalidad de tener una completa descripción del fenotipo. La revisión de libros (como el conocido y popular libro original de David Smith), revistas de la especialidad y programas de diagnóstico computarizado como son el London Dysmorphology Data Base (LDDB) o el Pictures Of Standard Syndromes and Undiagnosed Malformations (POSSUM) creado en Australia, son de uso frecuente en muchos países. Sin embargo, con el advenimiento de Internet, el acceso gratis a base de datos y publicaciones de excelente calidad como OMIM (Online Mendelian Inheritance in Man) creado por Victor A. McKusick en Johns Hopkins y actualizado diariamente, y GeneTest/GeneReviews, dirigido por Roberta Pagon, Universidad de Washington en Seattle, en los últimos años no hay prácticamente limitaciones para una exhaustiva búsqueda diagnóstica. Es interesante que en la actualidad, con la posibilidad de “googlear”, hay muchas familias que llegan a las consultas para una evaluación genética y ya han establecido o sospechado el diagnóstico correcto. Recientemente ha aparecido un programa, FACE2GENE, que usando Internet, permite el análisis dismorfológico facial gratuito usando fotos de pacientes. Sólo se requiere el registro previo del médico. El programa instantáneamente detecta y define los rasgos dismórficos y sugiere posibles diagnósticos.
PRINCIPALES HALLAZGOS SUGERENTES DE ABERRACIÓN CROMOSÓMICA
| 1)Retrasoorestriccióndelcrecimientointrauterino/Recién nacido pequeño para la edad gestacional |
| 2) Retraso del crecimiento postnatal / Falla para ganar peso |
| 3) Retraso del desarrollo psicomotor / Retraso intelectual |
| 4) Presencia de malformaciones congénitas |
| 5) Presencia de rasgos dismórficos |
| 6) Alteraciones neurológicas |
| 7) Anomalías dermatoglíficas* |
* En la reunión anual de la sociedad de genética Humana americana de 1997, lacassie y stahls presentaron cómo la ausencia de los triradios basales a, b, ó d, en presencia de varios de los hallazgos enumerados en esta tabla, indican una chance de aproximadamente 50% de encontrar una pequeña anomalía cromosómica que pudiera no haber sido detectada incluso con un estudio cromosómico de alta resolución (550 bandas o más); y en la reunión anual de 2009, lacassie, myrtle y sathyamoorthi, presentaron el trabajo “hand Dysmorphology: Dermatoglyphics – a neglected clue to suspect microdeletions.” En este estudio se reportó un 36% de anormalidades versus un 7% de la literatura. la sensibilidad y especifi de los rasgos estudiados fue la siguiente: ausencia de triradio axial (0.05 y 0.97); presilla distal en zonas interdigitales 2, 3 y 4 simultáneamente (0.07 y 0.98); ausencia del triradio “d” (0.11 y 0.92); triradio axial distal (t”) (0.17 y 0.89); recuento total de surcos digitales muy alto o muy bajo (0.21 y 0.86); pliegues palmares anormales (032. y 0.81); terminación de líneas principales muy transversales o verticales (0.32 y 0.75). con la presencia de más de 4 de estos hallazgos 100% de los pacientes presentaban una alteración. Este estudio no incluyó pacientes con síndrome de down, trisomías 13 o 18 fácilmente reconocibles clínicamente y que tienen dermatoglifos característicos que permiten su diagnóstico.
A pesar de realizar un buena y completa evaluación clínica, no siempre se logra establecer el diagnóstico. Esto es debido a que pueden existir diversos problemas.
PROBLEMAS PARA ESTABLECER EL DIAGNÓSTICOSi consideramos que muchísimas enfermedades genéticas y síndromes son raros; que no existen rasgos específicos o patognomónicos que permitan un diagnóstico directo; que existe gran heterogeneidad genética y variabilidad clínica; que las manifestaciones pueden aparecer a lo largo de la vida; que los nuevos exámenes moleculares además de caros tienen limitaciones, etc., es posible entender que establecer el diagnóstico certero no sea fácil. Además, hay una serie de otras situaciones que dificultan establecer el diagnóstico etiológico. En 1994 el autor fue invitado al Congreso Latinoamericano de Genética Humana en México. Allí presentó una clasificación de problemas para determinar un diagnóstico. En esta primera clasificación reconocimos problemas debidos al observador, al paciente, a la enfermedad genética y al ambiente. Sin embargo, en 2009 con los avances de los exámenes genéticos, agregamos un quinto grupo de problemas que son debidos a la informática. Es muy importante que los pediatras y médicos generales estén conscientes de la existencia de estos diferentes problemas para establecer el diagnóstico.
El primer grupo de problemas es debido al observador. No es infrecuente que se cometan más errores por evaluación incompleta que por falta de conocimiento. Con frecuencia el paciente no ha tenido un examen completo. Hay dos áreas del cuerpo que habitualmente proporcionan las claves diagnósticas más importantes: la cara y las manos. Sin embargo, con frecuencia, la evaluación de las extremidades se limita a observar los pliegues palmares de flexión ignorando otros elementos diagnósticos. Frecuentemente tampoco es examinada el área de los genitales. Otro problema común es la subjetividad. Por ejemplo, con frecuencia se reportan orejas o pabellones auriculares de inserción baja, sin embargo, el examen objetivo muestra que éstas no están bajas sino posteriormente rotadas o son displásticas. Indudablemente la capacidad del observador está directamente relacionada a su conocimiento y experiencia.
El segundo grupo de problemas de diagnóstico se debe al paciente o a la familia. Ocasionalmente hay pacientes que proporcionan información incompleta o equívoca. Debemos estar conscientes de la posibilidad de paternidad falsa. Es importante también evaluar la presencia de dismorfias en otros miembros de la familia. Con frecuencia los padres tratan de justificar los diferentes rasgos dismórficos del paciente como rasgos familiares. Ocasionalmente los pacientes pueden presentar, en coincidencia, dos o más condiciones genéticas de forma simultánea y esto indudablemente dificulta el diagnóstico. Históricamente hemos considerado en este grupo de problemas a los mortinatos o mortineonatos. Sin embargo, este grupo de pacientes en la actualidad puede no constituir un mayor problema de diagnóstico si se tiene la precaución de extraer ADN para futuros estudios genéticos, si fuesen necesarios.
El tercer grupo de problemas de diagnóstico es debido a la enfermedad genética. Este problema ha sido el más prevalente e importante en la literatura médica en los últimos sesenta años. La heterogeneidad genética existe en muchísimas condiciones. Diferentes genes, a veces con diferentes patrones de herencia, pueden producir un fenotipo similar. En la actualidad reconocemos la importancia de la interacción entre genes así como el fenómeno epigenético. Por epigenética se entiende el estudio de cambios heredables en la expresión génica o en el fenotipo celular causado por diferentes mecanismos sin cambiar la secuencia de ADN. Ejemplo de tales cambios podrían ser metilación del ADN, deacetilación de histonas, etc., que suprimen la expresión del gen sin alterar su secuencia. En la actualidad se entiende mejor el pleiotropismo, o cómo un gen puede afectar diferentes estructuras en el organismo. Esto también tiene que ver con la variabilidad clínica. No todos los síndromes presentan el cuadro característico. Abiotropismo es el término que indica que no todas las enfermedades genéticas son congénitas, es decir son evidentes al nacer; algunas pueden aparecer a lo largo de la vida ya sea a edades específicas o variables. Por otra parte, condiciones ambientales pueden simular herencia mendeliana, simulando una herencia autosómica dominante o recesiva. Como se mencionara, en los últimos treinta años se ha reconocido la importancia de patrones de herencia no tradicionales. También necesitamos estar conscientes de la posibilidad de estar frente a un mosaicismo gonadal que podría explicar por qué algunos padres normales pueden tener dos o más hijos afectados con la misma enfermedad autosómica dominante.
El cuarto grupo de problemas de diagnóstico es debido al ambiente. Esta idea nació hace más de treinta años trabajando en el INTA, especialmente en la Unidad Metabólica y en el Centro Normativo Macul de CONIN. Ahí reconocimos la existencia de muchas enfermedades genéticas que estaban ocultas bajo el diagnóstico de desnutrición. Cuando familias de buen nivel socioeconómico tienen un niño que presenta retraso del crecimiento o del desarrollo psicomotor, el pediatra rápidamente refiere el niño al neurólogo, genetista u otro especialista; mientras que cuando el mismo paciente nace en un ambiente deprivado, el retraso se atribuye a factores nutricionales o ambientales. En este grupo incluimos también limitaciones para realizar algunos estudios, especialmente los nuevos estudios moleculares. Hay muchos exámenes que se realizan sólo en algunos laboratorios de Estados Unidos, Europa o algún otro país. También es importante mencionar cómo el alto costo de algunos de estos exámenes es, con frecuencia, un factor limitante para establecer un diagnóstico etiológico.
El quinto grupo de problema de diagnóstico es el debido a la informática; es importante considerar que, a pesar de los avances en este campo, aún existen problemas con la interpretación de algunos resultados. El incompleto conocimiento de la correlación entre fenotipo y genotipo es un problema práctico con el cual en la actualidad nos enfrentamos a diario. Esperamos que con la experiencia que rápidamente se está acumulando y que debiera darse a conocer en congresos y publicaciones de la especialidad, esta situación mejore, permita establecer un alto porcentaje de diagnósticos etiológicos y deje de constituir un problema de diagnóstico como es en la actualidad.
Artículo dedicado al destacado científico, pediatra y visionario, profesor, jefe, mentor y amigo, Dr. Fernando Mönckeberg Barros, quien en 1966, hace casi 50 años, me mostró el futuro de la pediatría, la importancia de la genética y cómo el estudio de las manos podía ayudar a establecer ciertos diagnósticos. Agradezco al Dr. Mönckeberg el haberme señalado un camino nuevo, extraordinariamente interesante, lleno de desafíos pero pleno de realizaciones.
Agradecimiento: Agradezco a Paulette Lacassie Silva la transcripción de esta publicación.
El autor declara no tener conflictos de interés, en relación a este artículo.