



El impacto que representan las afecciones genéticas en las unidades de cuidado intensivo neonatal (UCIN) en Chile no se conoce con certeza, aunque se acepta que es cuantioso y creciente. El desarrollo de nuevas técnicas que permiten investigar las bases genómicas de los fenotipos humanos y la considerable disminución de los costos de los estudios moleculares han impulsado el uso de técnicas de análisis genómico en la práctica clínica, especialmente en las áreas de medicina neonatal. El principal grupo detectado en la etapa de recién nacido son las malformaciones congénitas, las que se observan en 2–3% de los recién nacidos vivos, cardiopatías congénitas en 1% y los errores congénitos del metabolismo en 0,5%. Se estima además que las malformaciones congénitas son responsables de un 13% de los ingresos a las UCIN y de más de un tercio de la mortalidad neonatal. Las nuevas tecnologías disponibles para el estudio molecular de enfermedades genéticas permiten el diagnóstico de precisión en un número muy importante de los pacientes críticamente enfermos ingresados a UCIN. El diagnóstico certero en el tiempo adecuado muchas veces modifica el manejo y mejora el pronóstico.
En este artículo se revisan las diferentes metodologías diagnósticas actualmente disponibles, sus indicaciones y rendimiento en el estudio etiológico de recién nacidos en los que se sospecha una condición de origen genético.
In Chile, the real burden of genetic conditions in neonatal intensive care units is unknown but it is accepted that it is high and increasing. The development of new technologies that allow the study of the genetic bases of human phenotypes and the considerable decrease of the costs of molecular analyses have driven the use of genomics assays techniques in clinical practice, especially in the area of neonatal medicine. In the newborn period the principal group of genetic anomalies detected are congenital malformations, which are present in 2-3% of live births, followed by congenital heart defects in 1% and inborn errors of metabolism in 0,5%. It is considered that congenital malformations are responsible for 13% of neonatal care unit's incomes, and one third of neonatal mortality. New available technologies for molecular study of genetic diseases permit precise diagnoses in a considerable number of critically ill patients in neonatal care units. Many times, an accurate diagnosis performed at the right time modifies the management and prognosis of the disease.
In this report all different diagnostic methodologies available and their indications in the study of etiological diagnosis of newborns suspected to have a genetic condition are reviewed.
Las enfermedades genéticas y las malformaciones congénitas son la primera causa de mortalidad infantil en Chile1. Esto ocurre desde hace ya varios años en países desarrollados, es así como en EE.UU. el año 2013 estas afecciones fueron responsables de más del 20% de las muertes en menores de 1 año2. Así mismo, se estima que 6,4-14,4% de los recién nacidos en EE.UU. requieren ser ingresados a algún tipo de unidad de cuidado neonatal y que la mitad de estos ingresos no se relacionan con problemas asociados a prematuridad3.
Las afecciones genéticas, incluyendo las alteraciones monogénicas, las variaciones en el número de copias (CNVs) y las alteraciones cromosómicas son condiciones individualmente poco frecuentes pero que en conjunto afectan una proporción importante de la población general4,5. A la fecha existe caracterización molecular para 7.001 de las casi 8.000 afecciones genéticas identificadas y se han descrito 6.202 afecciones monogénicas, producidas por cambios en 4.526 genes6.
No se conoce exactamente el impacto que representan las afecciones genéticas en las UCIN en Chile, aunque se acepta que su impacto es importante y creciente. Cada vez con más frecuencia se reciben recién nacidos con diagnóstico prenatal confirmado y otros son diagnosticados en las primeras horas o días de vida cuando se trata de fenotipos reconocibles, sin embargo, muchos diagnósticos son difíciles de precisar7, y frente a la sospecha de una afección genética subyacente es importante conocer las herramientas disponibles para establecer un diagnóstico de precisión.
El campo de la genética médica ha ido progresivamente adquiriendo un rol importante en los estudios diagnósticos. El desarrollo de nuevas técnicas que permiten investigar las bases genómicas de los fenotipos humanos y la considerable disminución de los costos de los estudios moleculares han impulsado el uso de técnicas de análisis genómico en la práctica clínica, especialmente en las áreas de medicina neonatal, a pesar de que las enfermedades genéticas pueden manifestarse a cualquier edad8,9.
Sin embargo, antes de realizar cualquier estudio diagnóstico es fundamental la evaluación clínica exhaustiva, la que debe incluir un análisis detallado de la historia familiar. No existen recomendaciones específicas de que el estudio debe hacerse a cada paciente para descartar afecciones genéticas en la etapa neonatal. Incluso existe la propuesta, lejana a nuestra realidad, de realizar estudio de secuenciación completa de exoma o genoma, en todo recién nacido críticamente enfermo en el que se sospeche una afección genética subyacente10–12.
2EpidemiologíaComo se señaló, el real impacto de las afecciones genéticas en una UCIN no se conoce, pues muchas condiciones potencialmente genéticas con frecuencia no son estudiadas, no son evidentes o se expresan solo parcialmente durante el periodo de hospitalización. El principal grupo detectado en la etapa de recién nacido son las malformaciones congénitas, las que se observan en 2–3% de los recién nacidos vivos, cardiopatías congénitas en 1% y los errores congénitos del metabolismo en 0,5%13. Se estima además que las malformaciones congénitas son responsables de un 13% de los ingresos a las UCIN y de más de un tercio de la mortalidad neonatal14.
Se calcula que las malformaciones congénitas, las afecciones neuromusculares, el retraso del neurodesarrollo y la discapacidad intelectual en global, afectan a un 10% de los recién nacidos vivos y que alrededor de 20% de la mortalidad infantil en EE.UU. se debe a malformaciones congénitas y/o a anomalías cromosómicas13. Durante los últimos 30 años, en los países desarrollados, la mortalidad por defectos congénitos se ha mantenido relativamente estable, a diferencia de la importante reducción, cercana a 50%, que ha mostrado la tasa global de mortalidad infantil10. En Chile, en general, los números son muy similares1.
Un análisis más amplio de las tasas regionales realizado por la Organización Panamericana de la Salud, muestra que anualmente nacen en la Región de las Américas más de 15 millones de niños. De ellos, en 2017, aproximadamente 15 de cada 1.000 morirán antes de cumplir 1 año y 10 de cada 1.000 antes de cumplir un mes de vida. Las muertes neonatales estimadas (103.000) en 2017 ocurridas en América Latina y el Caribe representaron casi dos tercios (65,5%) de todas las muertes en el primer año de vida y 55% de todas las muertes de niños menores de 5 años. De las defunciones durante los primeros 28 días de vida, 1 de cada 5 se debe a defectos congénitos, lo que traducido a números absolutos representa casi 20.000 niños. Debido a los logros alcanzados en términos de reducción de otras causas de muerte prevenibles en este grupo de edad, la proporción de muertes neonatales causados por defectos congénitos aumentó de 16,2% a 22,3% entre los años 2000 y 2016. En términos generales, la mortalidad proporcional debida a defectos congénitos está incrementando con el paso del tiempo en la mayoría de los países. Sin embargo, no se aprecia un incremento claro de las tasas de mortalidad neonatal por esta causa, aun cuando cabe considerar que en general están bajando15.
3¿Qué pacientes deben ser estudiados?Cada vez con más frecuencia hay sospecha o diagnóstico prenatal confirmado de una afección genética en el periodo prenatal. La mejoría en la resolución de las imágenes de ultrasonido, la disponibilidad de resonancia magnética fetal y de estudios moleculares, ya sea como tamizaje a través del diagnóstico prenatal no invasivo (NIPT por sus siglas en inglés) o de certeza, a través de técnicas citogenéticas o moleculares, entregan cada vez información más certera al equipo de médicos tratantes permitiendo enfrentar de mejor manera el nacimiento del niño afectado. Sin embargo, aún persiste un número importante de casos en los que el equipo médico se ve enfrentado a la sospecha de una afección genética subyacente en el periodo neonatal inmediato o en los primeros días de vida.
Las situaciones en las que esto ocurre se señalan a continuación10.
- -
Recién nacido con malformaciones congénitas severas y cuyo fenotipo no es compatible con un síndrome conocido.
- -
Condiciones críticas con compromiso vital, de etiología no precisada (falla orgánica múltiple).
- -
Enfermedad grave de algún órgano vital, de patogenia desconocida y sin respuesta a las terapias estándares.
- -
Trastorno neurológico grave de origen desconocido.
- -
Falla metabólica de origen desconocido.
- -
Retraso severo del crecimiento intrauterino de origen desconocido.
- -
Otras condiciones agudas, graves, de origen desconocido.
Sin duda, la manifestación más conocida y llamativa de las afecciones genéticas son los síndromes de múltiples malformaciones congénitas, para los que el hombre desde siempre ha buscado una explicación. Estas explicaciones en las distintas épocas han pasado desde las maldiciones de los dioses, hibridación con animales, relación con fenómenos naturales como los eclipses, etc. En la actualidad es posible establecer el origen certero en un porcentaje muy alto de estos defectos.
Es importante mencionar también que no todos los defectos congénitos tienen una base genética y es así como siempre hay que tener presente, en el diagnóstico diferencial, la posibilidad de que el defecto sea secundario a la acción de un teratógeno o producto de una interferencia externa al desarrollo embrionario o fetal, como ocurre con las bridas amnióticas. También es importante tener presente que existen fenotipos muy característicos de un diagnóstico pero en los que el genotipo es altamente heterogéneo, por lo que es importante realizar un estudio genético molecular que incluya todos los eventuales genes involucrados, como ocurre, a modo de ejemplo, en el síndrome de Noonan, en el que se ha descrito asociación en al menos 9 genes: PTPN11 en 50% de los afectados, SOS1 en alrededor de 13%, RAF1 y RIT1 cada uno en 5%, KRAS en menos del 5% y otros como BRAF, LZTR1, MAP2K1 y NRAS en los que se han detectado mutaciones en menos del 1% de los casos con Síndrome de Noonan16. Sin embargo, el escenario más frecuente al que se ve enfrentado el genetista clínico es un fenotipo en el que no se reconoce elementos de una afección genética clásica y tampoco existe historia para plantear un origen no genético como una enfermedad materna. Frente a cada una de estas alternativas el enfrentamiento desde el punto de vista del estudio diagnóstico será diferente10.
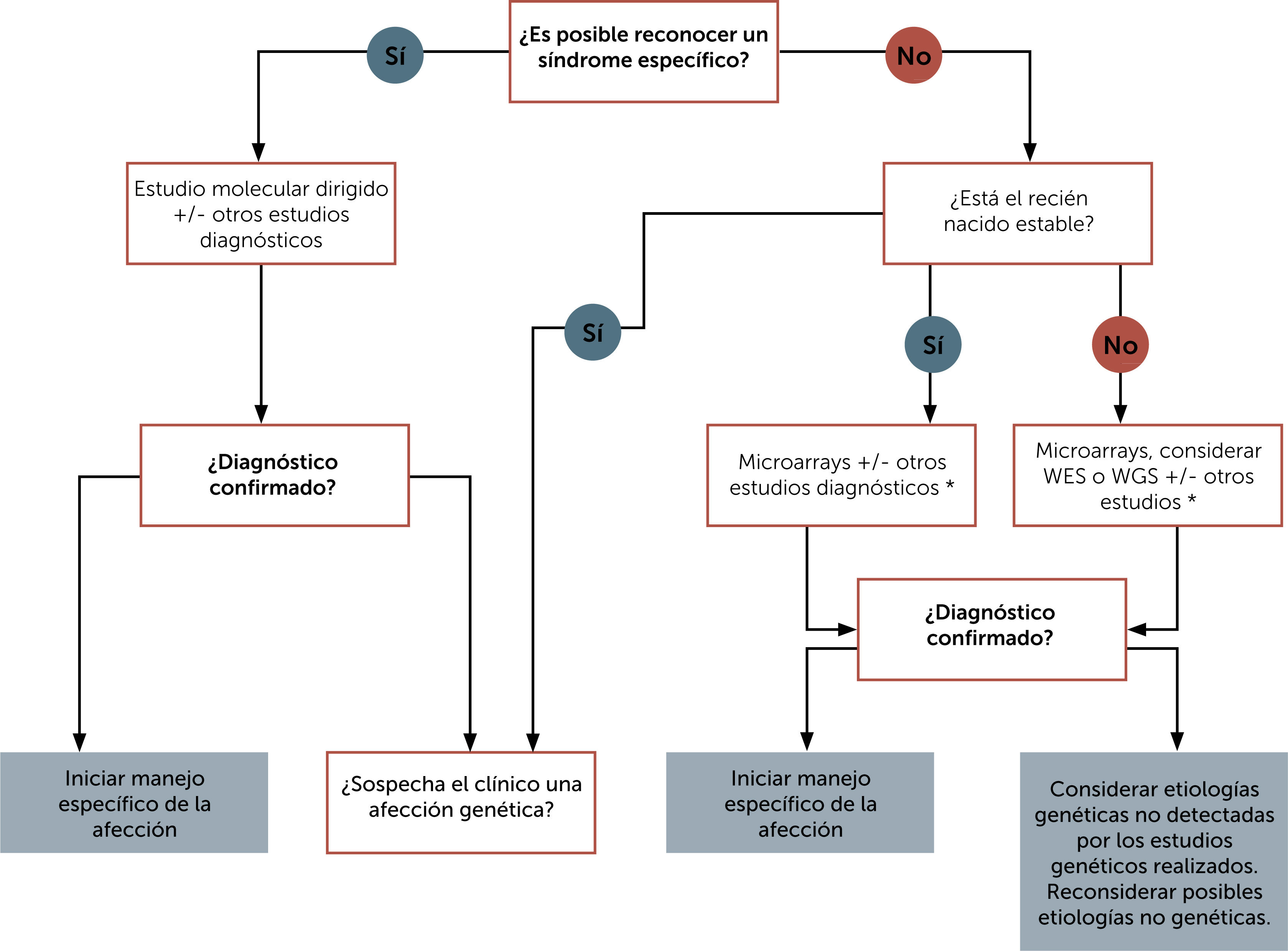
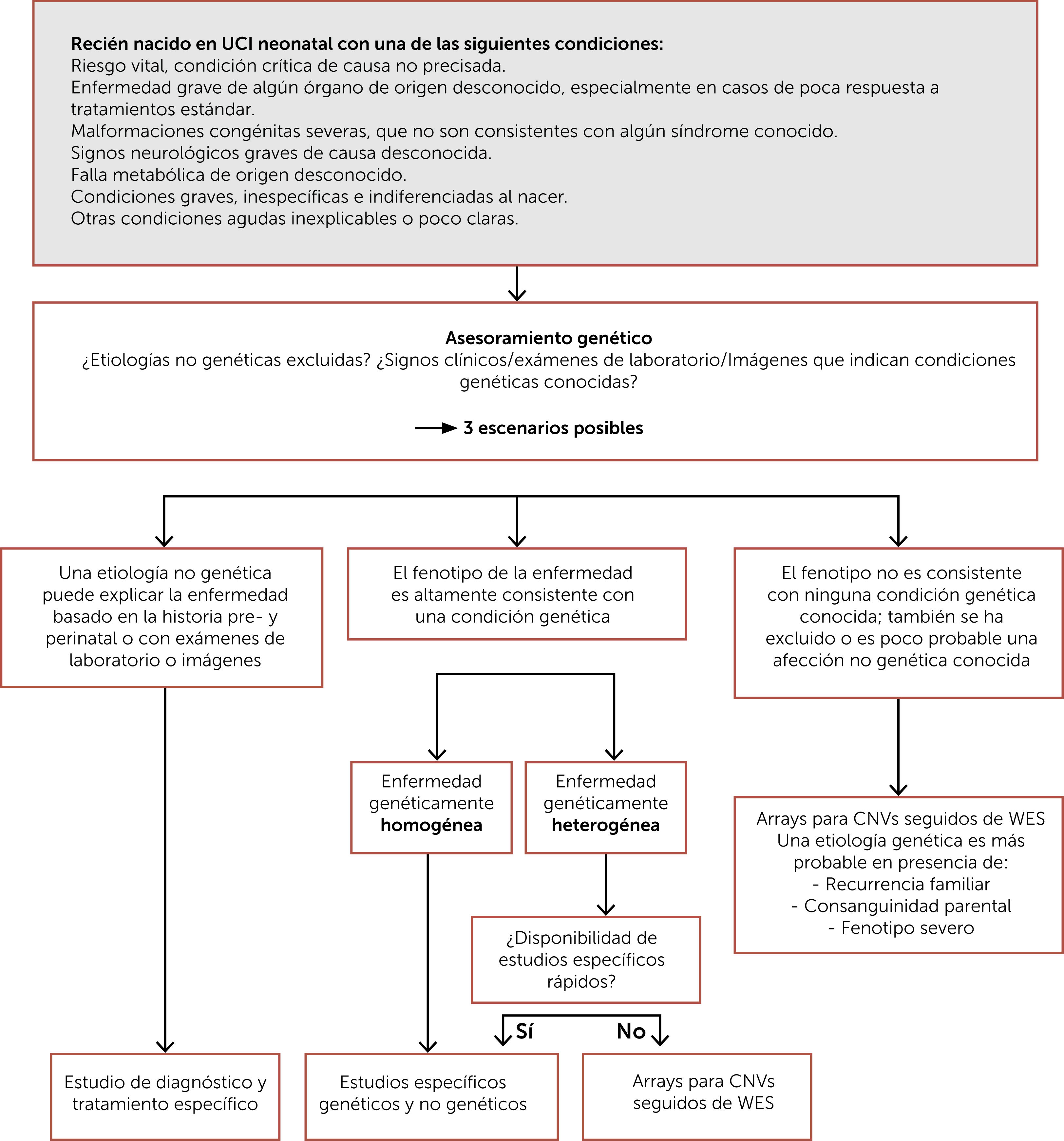
Contar con un diagnóstico de precisión es fundamental para la familia y para el equipo tratante, pues permite determinar la disponibilidad de tratamiento específico, la posibilidad de acceso a estudios clínicos, la importancia de evitar algunos agentes con eventual contraindicación, el retiro de terapias no requeridas, entre otros y a los padres les permite conocer el pronóstico, posibles complicaciones, unirse a agrupaciones de padres, tomar decisiones reproductivas, entre otros. En la Fig. 1 se muestra un esquema de enfrentamiento diagnóstico al recién nacido con malformaciones congénitas y en la Fig. 2 un esquema de enfrentamiento diagnóstico al recién nacido críticamente enfermo.
. Abreviaturas: WES: secuenciación completa de exones; WGS: secuenciación del genoma completo.")
Algoritmo de enfrentamiento diagnóstico del recién nacido con múltiples anomalías congénitas. Traducida de Carroll J. et al. (Ref.17). Abreviaturas: WES: secuenciación completa de exones; WGS: secuenciación del genoma completo.
. Abreviaturas: CNVs: variaciones en el número de copias; WES: secuenciación completa de exones.")
Algoritmo para el estudio diagnóstico del recién nacido críticamente enfermo. Traducida de Borghesi A et al. (Ref.10). Abreviaturas: CNVs: variaciones en el número de copias; WES: secuenciación completa de exones.
Todo lo anterior permitirá al equipo médico de las unidades neonatales mejorar el manejo clínico y secundariamente disminuir la morbimortalidad y también, en aquellos casos con pronóstico vital desfavorable, y en conjunto con una familia bien informada, tomar decisiones tendientes a adecuar el esfuerzo terapéutico.
4¿Cuáles son los exámenes disponibles y cuando solicitarlos?Los avances en genética molecular y la disponibilidad, cada vez a costos más accesibles, de las nuevas tecnologías permiten cada día acceder a ellas con mayor facilidad. Sin embargo, aun en muchas oportunidades son los estudios más clásicos los que nos permiten realizar diagnósticos certeros.
A continuación, se describen los exámenes disponibles y su utilidad en pacientes ingresados a una unidad de cuidados críticos neonatales, y en los que se sospecha una afección genética:
4.1CariogramaEs el primer estudio citogenético del que se dispuso y hasta hoy se mantiene como el examen de elección para el diagnóstico de aneuploidías. Es también el único examen que permite la detección certera de alteraciones cromosómicas balanceadas.
Permite también la detección de deleciones y duplicaciones de hasta 5–10 Mb, dependiendo de la resolución del bandeo y de la ubicación de la alteración dentro del cromosoma y de rearreglos estructurales balanceados y no balanceados. El cariograma es habitualmente el primer estudio solicitado en recién nacidos con múltiples anomalías congénitas y como se señaló es el estudio de elección frente a las sospecha de aneuploidías como síndrome de Down (trisomía 21), síndrome de Edwards (trisomía 18), síndrome de Patau (trisomía 13) y síndrome de Turner (monosomía X o 45, X), permitiendo no solo establecer el diagnóstico sino que también identificar si la alteración es producto de una no disyunción o si se trata de una translocación no balanceada heredada a partir de una alteración balanceada de uno de los progenitores. El cariograma permite también detectar mosaicos (dos o más líneas celulares con complementos cromosómicos diferentes) y por supuesto también los complementos cromosómicos poliploides (triploidías, tetraploidías, entre otros). La principal desventaja del cariograma es su limitada resolución ya que no permite detectar alteraciones menores a 5-10 Mb así como tampoco permite detectar otras alteraciones como las disomías uniparentales17.
Indicaciones de estudio cromosómico en recién nacidos:
- •
Recién nacidos con un síndrome cromosómico clásico.
- •
Recién nacidos con múltiples malformaciones congénitas (más de dos no relacionadas).
- •
Recién nacidos con trastornos de la diferenciación sexual.
- •
Hijos de padres portadores de alteraciones cromosómicas.
- •
Sospecha prenatal de aneuploidía.
Es una técnica de citogenética molecular que marca secuencias nucleotídicas a través de sondas unidas a un fluorocromo. Esta técnica permitió un gran avance en el campo de la citogenética ya que detecta deleciones y duplicaciones submicroscópicas en un rango de 50–200 Kb18. Se usa habitualmente para el diagnóstico rápido de aneuploidías y de síndromes de microdeleción (por ej.: del22q11.2) y para el diagnóstico de sexo genético en recién nacidos con trastornos de la diferenciación sexual (sondas para gen SRY y para cromosoma X). Las ventajas de esta técnica son: de bajo costo, capacidad de detección de alteraciones pequeñas y rapidez de ejecución ya que no requiere cultivos celulares. Su mayor desventaja es la sospecha diagnóstica que debe ser precisa puesto que se requiere del uso de sondas específicas para cada diagnóstico. También es importante señalar que no detecta alteraciones de tamaños menores a 50 Kb, y puede llevar a un subdiagnóstico.
Para su ejecución se utilizan diferentes tipos de sondas dependiendo del diagnóstico de sospecha:
- -
Centroméricas: sirven para detectar alteraciones numéricas. Se pueden realizar en núcleos.
- -
Sondas de secuencia única (locus específico), hibridan con secuencias cromosómicas concretas. Se utilizan para estudio de microdeleciones o duplicaciones. Requieren células en división.
- -
Sondas de pintado cromosómico: contienen una librería de secuencias genómicas que abarcan todo un cromosoma o una región específica. Permiten detectar alteraciones estructurales o numéricas. Requieren células en división. No son de uso clínico habitual.
Indicaciones de estudio de FISH en recién nacidos
- •
Diagnóstico rápido de aneuploidías.
- •
Estudio de microdeleciones.
- •
Asignación de sexo genético en recién nacidos con trastorno de la diferenciación sexual.
La técnica de MLPA (Multiplex Ligation-dependent Probe
Amplification) permite el estudio de las variaciones del número de copias de genes (CNV) asociados a enfermedades. Existen kits específicos para cada enfermedad que se desee analizar, por ello, cada kit contiene una mezcla de sondas especificas diseñadas de forma que contienen una región específica y otra región universal para amplificarlas todas juntas en una sola reacción de PCR. Si no hay alteraciones en las secuencias, las sondas se ligan, se amplifican, se separan por electroforesis capilar y se analizan los datos en un software especial. De esta forma se pueden detectar cambios en el número de copias desde cromosomas completos hasta exones individuales. Esta técnica también se usa para detectar cambios en los patrones de metilación del ADN (MS-MLPA) y es lo suficientemente sensible como para discriminar aberraciones en genes causantes de enfermedades que tienen pseudogenes (genes muy similares). Por lo tanto, permite detectar aneuploidías, deleciones, duplicaciones y disomías uniparentales. También permite determinar el origen parental de los fragmentos, pero no permite detectar mosaicos bajos, triploidías femeninas ni alteraciones estructurales balanceadas.
6.1Hibridación genómica comparativa (CGH array)Esta técnica permite detectar variaciones en el número de copias del genoma, es decir, detecta la cantidad de información genómica de un individuo. Es por lo tanto la técnica de elección para el estudio de deleciones o duplicaciones submicroscópicas y permite además detectar aneuploidías. Las técnicas de hibridación comparativa de genoma pueden ser complementadas con el análisis de polimorfismos de nucleótido único (SNP-array), permitiendo un análisis más fino de las variaciones del genoma. Consiste en marcar con fluorósforos diferentes el ADN genómico en estudio y el de un individuo normal y cohibridarlos con cromosomas metafásicos normales, para posteriormente analizarlos con un software específico que permite detectar cambios genómicos, ganancias o pérdidas, con una sensibilidad de 10-20 Mb.
El CGH array fue la primera técnica de microarray utilizada clínicamente y usa sondas que hibridan en diferentes regiones a lo largo del genoma permitiendo definir si hay ganancias o pérdidas. Al agregar SNPs es posible determinar la heterocigocidad (origen parental de cada región) permitiendo también determinar disomías uniparentales y los eventuales ancestros comunes (mientras mayor es la pérdida de heterocigocidad, mayor es la posibilidad de ancestros comunes). Dependiendo de la plataforma que se utilice, el tipo y la densidad del array utilizado (60, 180 o 400K), cambia la resolución. A diferencia del FISH, el CGH permite el análisis simultaneo de millones de loci, no requiere sospecha clínica específica y puede detectar deleciones y duplicaciones de mucho menor tamaño, permitiendo mayor precisión en la determinación de los puntos de quiebre y de los genes involucrados en la alteración.
En resumen, el CGH array es una técnica de mayor rendimiento que el cariograma y el FISH en el estudio de patologías genéticas ya que permite la detección, además de las aneuploidías, de alteraciones submicroscópicas (microdeleciones y microduplicaciones) y también de alteraciones cromosómicas desbalanceadas. No permite detectar translocaciones balanceadas ni mosaicos bajos. La interpretación de los resultados requiere de bases de datos extensas de población normal y afectada.
Los resultados se informan como: patogénicos, probablemente patogénicos, variantes de significado incierto (VUS), probablemente benignos o benignos. Los cambios definidos como probablemente patogénicos deben ser analizados a la luz del fenotipo y las variantes de significado incierto, deben ser además ser correlacionadas con el genotipo de los progenitores.
Indicaciones del uso de CGH- array en recién nacidos
- •
Recién nacidos con múltiples malformaciones congénitas (con cariotipo normal).
- •
Recién nacidos con síndromes dismórficos de etiología no precisada.
- •
Recién nacidos en los que el estudio cromosómico muestra cromosomas marcadores.
- •
Recién nacido con retraso de crecimiento intrauterino, de etiología no precisada, y con cariotipo normal.
La técnica de PCR en tiempo real (RT-PCR) permite la amplificación de un segmento de ADN que es flanqueado por dos partidores. Con esto se logra obtener muchas copias de un gen y compararla con un gen normal. Es una técnica sensible, rápida y económica. Su principal aplicación clínica es la genotipificación de agentes infecciosos pero además es de gran utilidad en la detección de mutaciones puntuales responsables de una enfermedad genética, con poca o nula variabilidad genotípica como ocurre en la acondroplasia en la que más del 98% de los pacientes presenta una única mutación en el gen FGFR3 (c.1138G>A; p.Gly380Arg)19,20.
6.3Southern BlotEs un método que permite detectar la presencia de una secuencia específica, en una mezcla compleja de ADN. Su principal aplicación en clínica es que permite determinar la expansión de tripletes en algunas enfermedades genéticas (Corea de Huntington, Síndrome de X-frágil). Su utilidad en neonatología estaría solo en los casos en que existe un riesgo aumentado, por antecedentes familiares, de presentar una afección de este tipo, pero principalmente en el diagnóstico de distrofía miotónica, afección producida por un aumento de repetidos CTG en el gen DMPK ubicado en 19q13 y que en el periodo neonatal se manifiesta por un cuadro grave de compromiso neurológico.
6.4Secuenciación genómicaEs un conjunto de métodos y técnicas bioquímicas cuya finalidad es la determinación del orden de los nucleótidos (A,C,G,T) en un fragmento de ADN. Es una técnica que permite el diagnóstico de un gran número de enfermedades monogénicas y es la técnica de elección para identificar mutaciones puntuales.
Existen dos técnicas básicas de secuenciación: secuenciación Sanger y la secuenciación masiva o secuenciación de nueva generación (NGS).
6.5Secuenciación SangerDescrita en 1975, es la técnica de secuenciación más utilizada para el estudio de segmentos específicos de ADN o de genes pequeños. Es una técnica muy confiable, aunque su ejecución es lenta por lo que no es la técnica de elección si se requiere secuenciar varios genes o un gen grande en el que se han descrito varias mutaciones. Se utiliza siempre para corroborar una alteración detectada por secuenciación masiva. Cuando se solicita este estudio es importante saber qué es lo que ofrece cada laboratorio ya que algunos solo estudiarán las variantes patogénicas más frecuentes y otros harán la secuenciación completa del gen.
Indicación de secuenciación Sanger en neonatología
- -
Recién nacido en el que se sospecha fuertemente una afección genética de baja heterogeneidad genotípica.
- -
Recién nacido con riesgo de presentar una afección genética monogénica conocida, por antecedentes familiares. Si se conoce la alteración molecular subyacente en la familia se solicita ese estudio puntual.
Es una técnica que permite secuenciar en un solo ensayo un gran número de genes, simplificando la metodología de trabajo y aumentando la capacidad y velocidad de secuenciación. Involucra la secuenciación de millones de pequeños fragmentos de ADN en forma simultánea, permitiendo que cada base a lo largo del genoma sea secuenciada múltiples veces. Esto permite analizar, a un costo menor, un gran número de muestras o muestras que requieren estudio de un gran número de genes. Esta metodología también permite la construcción de paneles a medida (se solicita el estudio de diferentes genes de acuerdo a las características fenotípicas del paciente).
Al igual que en los estudios con CGH, la patogenicidad de las variantes detectadas se informa en una escala de 5 puntos: patogénica, probablemente patogénica, variable de significado incierto (VUS), probablemente benigna y benigna. Las dos primeras son consideradas diagnósticas, las dos últimas habitualmente no se reportan y se consideran variantes polimórficas y las variantes de significado incierto requieren análisis en relación al fenotipo y estudio de los padres para determinar si son heredadas o de novo, si son heredadas desde un progenitor sano, es muy poco probable que sean responsables de enfermedad17.
Es la tecnología más apropiada para el diagnóstico de enfermedades con heterogeneidad génica y para la identificación de nuevos genes asociados a enfermedades genéticas.
6.7Paneles de secuenciación masivaLos paneles genéticos se han desarrollado para evaluar un grupo seleccionado de genes relacionados con un fenotipo en particular, es decir para evaluar fenotipos con heterogeneidad genética. En neonatología específicamente, los más utilizados son los asociados a epilepsia, miocardiopatías, hiperinsulinismo y colestasia21. Existen también otros paneles más amplios que permiten estudiar fenotipos menos específicos y, como se señaló, también se pueden diseñar paneles a medida. Sin embargo, a medida que disminuyen los costos de las tecnologías involucradas, cada vez se solicitan estudios más amplios como la secuenciación completa de exoma (WES) o genoma (WGS).
6.8Secuenciación completa de exoma (WES)En la actualidad la mayor utilización de la metodología de secuenciación masiva es la secuenciación completa de exoma (WES)22, la que consiste en la secuenciación simultanea de todas las regiones codificantes del genoma, que son los exones y unas pocas variantes intrónicas cercanas a los exones; permite identificar variaciones de nucleótidos únicos (SNVs) y pequeñas inserciones y deleciones (InDels) y se usa principalmente para el diagnóstico de afecciones monogénicas.
El genoma humano, formado por casi unos 20.000 genes, contiene cerca de 180.000 exones (2% del genoma). Los exones son secuencias cortas de ADN que se traducen en proteínas, por eso se conocen como regiones codificantes y contienen alrededor del 85% de las mutaciones responsables de enfermedad. Para hacer una mejor interpretación de los resultados de un estudio con WES, lo ideal es hacer análisis del trío (caso índice y ambos progenitores).
Estudios realizados en unidades neonatales en pacientes con sospecha de enfermedades genéticas en los que se ha realizado WES muestran tasas diagnósticas de entre 20 y 60% dependiendo de lo estricto de los criterios de selección23,24.
Por otra parte, WES tiene una mayor tasa de detección diagnóstica que los paneles ya que permite identificar cambios responsables de enfermedad en genes no incluidos en el panel además de detectar >98% de las variantes patogénicas de los genes incluidos en los paneles25. Por otra parte, como WES no está circunscrito a genes conocidos como responsables de enfermedad, permite el descubrimiento de nuevos genes y el re-análisis a la luz del conocimiento nuevo26. Finalmente, el uso de WES disminuye el tiempo requerido para tener un diagnóstico definitivo, lo que determina una muy buena relación costo-efectividad.
6.9Secuenciación completa de genoma (WGS)WGS utiliza la tecnología de secuenciación masiva para secuenciar con una cobertura uniforme, todo el ADN nuclear (exones e intrones) y el ADN mitocondrial. Se solicita este estudio en pacientes con sospecha de afección genética y con CGH y WES normales. La utilidad diagnóstica de WGS en las unidades de cuidado crítico neonatal se ha evaluado en varias publicaciones que muestran un rango de positividad que fluctúa entre 19 y 57%17,27. Debido a la cobertura más uniforme y completa, WGS es superior al WES si se considera la capacidad de evaluar el número de copias y variantes estructurales28. Al igual que WES tiene limitaciones: dificultad en secuenciar algunas regiones (secuencia 90% del genoma), en mapear los pseudogenes y en la detección de trinuceótidos repetidos.
Existen publicaciones recientes que proponen el uso de técnicas rápidas de secuenciación de genoma (rWGS) para el estudio diagnóstico de recién nacidos críticamente enfermos en los que sospecha una afección genética29 y también de cribado genómico neonatal30. Ambas situaciones requieren aun de mayor experiencia y disminución del costo de los estudios para ser aplicados en forma masiva.
Indicaciones de estudio con técnicas de secuenciación masiva en neonatología9.
- -
Sospecha de una afección genética, con un fenotipo inespecífico y no consistente con un síndrome conocido.
- -
Presencia de una condición consistente con una enfermedad genética conocida y la que se sabe es genéticamente heterogénea en su origen.
- -
Sospecha de una condición genética en un recién nacido críticamente enfermo y en quien el diagnóstico rápido es relevante para tomar decisiones de manejo.
Es importante señalar también que, si bien los avances en las tecnologías diagnósticas de afecciones genéticas son notables, existen una serie de dificultades en el periodo neonatal que dificultan el diagnóstico:
- –
Variabilidad fenotípica (penetrancia incompleta, expresividad variable).
- –
Heterogeneidad genética, aun en afecciones consideradas homogéneas y con fenotipos clínicamente reconocibles, lo que complica el análisis diagnóstico.
- –
Bajo índice de sospecha de afecciones genéticas en recién nacidos porque el fenotipo no siempre se expresa completo en las primeras etapas de la vida o el fenotipo se enmascara en pacientes en condiciones críticas.
Las nuevas tecnologías disponibles para el estudio molecular de enfermedades genéticas permiten el diagnóstico de precisión en un número muy importante de los pacientes críticamente enfermos ingresados a unidades de cuidados neonatales. El diagnóstico certero en el tiempo adecuado muchas veces modifica el manejo y mejora el pronóstico.
Cuando el fenotipo es altamente específico, el enfrentamiento puede ser dirigido (cariograma, FISH), pero cuando el fenotipo es inespecífico o sugerente de una afección genéticamente heterogénea, es necesario realizar estudios más amplios como CGH array, paneles, WES o WGS. Si bien no existen guías clínicas en relación con cuando pedir estos estudios, cada día se solicitan con más frecuencia y en forma más temprana, lo que ha permitido mejorar la oportunidad de realizar diagnósticos de precisión, conociendo los beneficios y limitaciones de cada uno de estos estudios.
En el caso de los defectos congénitos, la tasa de diagnóstico de WES es 20-25%, si el análisis se restringe a genes conocidos responsables de enfermedad y puede aumentar hasta cerca de 70% cuando se hacen estudios más amplios, con una mejor descripción fenotípica (incluyendo exámenes de laboratorio e imágenes) y además se estudia el trío (caso índice más progenitores). El análisis de la inmensa cantidad de datos obtenidos por WES o WGS requiere del análisis exhaustivo realizado por expertos bioinformáticos, quienes apoyados por clínicos, entregaran informes que serán más certeros en la medida que se haya aportado mayor información clínica9.
Declaración de conflicto de interésLa autora declara no tener conflicto de interés.