






Han pasado 35 años desde la aparición de los primeros casos de pacientes diagnosticados con VIH, de ahí en adelante la industria farmacéutica ha investigado e invertido millones de pesos en la búsqueda de terapias altamente efectivas (TARGA o HAART en inglés), con menos comprimidos, idealmente en dosis única y de baja toxicidad. La llegada de la terapia antiretroviral (ARV) y la combinación de diferentes pautas de estos, ha modificado la evolución natural de la infección, convirtiéndola en una patología crónica y reduciendo su morbi-mortalidad.
El uso combinado de diferentes fármacos antirretrovirales (ARV) ha permitido controlar la replicación viral, disminuir la activación inmune y preservar y/o restaurar el sistema inmune en gran parte de los pacientes, aproximando la esperanza de vida cada vez más a la de la población general. No obstante y debido a la imposibilidad actual de erradicar los reservorios del virus, es necesario mantener el tratamiento antirretroviral de por vida. Por otro lado, la complejidad de algunos esquemas y sus efectos adversos dificultan la adherencia, aumentando el riesgo de desarrollo de resistencias a fármacos en aquellos pacientes donde la adherencia es un problema.
El Ministerio de Salud de Chile (MINSAL) y la Organización Panamericana de la Salud OPS/OMS, se comprometieron a implementar acciones para cumplir con la meta 90-90-90 al año 2020 que consiste en aumentar al 90% la proporción de personas con VIH que conocen su diagnóstico, a incrementar al 90% aquellas bajo tratamiento antirretroviral, y a que el 90% bajo tratamiento tenga carga viral suprimida (indetectable).
En la actualidad existen disponibles 6 familias o clases que pueden ser agrupadas de acuerdo a su mecanismo de acción: 1) inhibidores de la transcriptasa inversa análogos de nucleósidos/nucleótidos (INTR) (abacavir, didanosina, emtricitabina, lamivudina, zidovudina y tenofovir), 2) inhibidores de la transcriptasa inversa no análogos de nucleósidos (INNTR) (efavirenz, nevirapina, etravirina y rilpivirina), 3) inhibidores de la proteasa (IP) (atazanavir, darunavir, fosamprenavir, lopinavir, ritonavir, saquinavir), 4) inhibidores de la entrada (enfuvirtide o T-20), 5) Antagonista de correceptores CCR5 (maraviroc) y 6) inhibidores de la integrasa (INSTI) (raltegravir, elvitegravir, dolutegravir). Todos ellos se encuentran indicados en el tratamiento de la infección por el VIH-1, en combinación con otros ARV. La mayoría de ellos han demostrado ser también activos frente al VIH-2 (excepto los inhibidores de la transcriptasa inversa no análogos de nucleósidos, enfuvirtide y maraviroc) y algunos son activos frente al virus de la hepatitis B (lamivudina, emtricitabina y tenofovir). Habitualmente se emplean combinaciones de tres fármacos activos, de acuerdo a las características individuales de cada paciente, del escenario clínico (naive, rescate, simplificación) y de la posibilidad de resistencia frente a algunos fármacos. Sin embargo, actualmente se están estudiando combinaciones de dos fármacos e incluso existen ya, datos de eficacia de la administración de un IP en monoterapia en determinadas circunstancias, o uso de IP + 1 INTR con resultados de eficacia similar al uso de 3 drogas en pacientes naive.
Por temas prácticos en esta revisión, se detallarán solo aquellos antiretrovirales que se encuentran vigentes, excluyendo aquellos que han sido discontinuados del mercado farmacéutico. Se analizarán las principales características farmacológicas de las diferentes familias de ARV, con sus ajustes de dosis en insuficiencia renal, su farmacocinética, mecanismos de acción, efectos adversos, interacciones contraindicadas, y el impacto de la terapia antiretroviral sobre el riñón, hígado y el perfil lipídico.
The advent of antiretroviral therapy (ART) and the combination of different patterns of these, has changed the natural history of infection turning it into a chronic disease and reducing morbidity and mortality.
Currently the combined use of different antiretroviral (ARV) enabled control viral replication, reduce immune activation and preserve and/or restore the immune system in most patients, bringing life expectancy increasingly to the general population. However, due to the current inability to eradicate the virus reservoirs it is necessary to maintain antiretroviral therapy for life. Moreover, the complexity of some schemes and their adverse effects hinder adhesion, increasing the risk of developing drug resistance in patients where adhesion is a problem.
Currently there are available 6 families or classes that can be grouped according to their mechanism of action: 1.- inhibitors analogue reverse transcriptase nucleoside/nucleotide (INTR) (abacavir, didanosine, emtricitabine, stavudine, lamivudine, zidovudine and tenofovir) 2.- inhibitors not analogous nucleoside reverse transcriptase inhibitors (NNRTIs) (efavirenz, nevirapine, etravirine and rilpivirine), 3.- protease inhibitors (PI) (atazanavir, darunavir, fosamprenavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, tipranavir) 4.- inhibitors input (enfuvirtide or T-20), 5.- CCR5 Antagonist (maraviroc) and 6.- integrase inhibitors (INSTI) (raltegravir, elvitegravir, dolutegravir). They are indicated in the treatment of HIV-1, in combination with other ARV. Most of them have also proven to be active against HIV-2 (except inhibitors non nucleoside reverse transcriptase nucleoside enfuvirtide and maraviroc) and some are active against hepatitis B (lamivudine, emtricitabine and tenofovir). Usually combinations of three active drugs, according to the individual characteristics of each patient, the clinical scenario (naive, rescue, simplification) and the possibility of resistance to certain drugs are used. However, currently being studied combinations of two drugs and even already exist, data management effectiveness of an IP monotherapy in certain circumstances, or use of IP + 1 NRTI with results similar efficacy to the use of 3 drugs in patients naive.
For practical issues in this review, detailing only those antiretrovirals currently in force, excluding those that have been discontinued in the pharmaceutical market. the main pharmacological characteristics of different families of ARVs will be analyzed, with dose adjustments in renal insufficiency, pharmacokinetics, mechanisms of action, adverse effects, contra interactions, drug delivery via nasogastric tube (NGT) and the impact of antiretroviral therapy on the kidney, liver and lipid profile.
Esta clase farmacológica es la más antigua y su uso asociado (con dos drogas) constituye el backbone o esqueleto del tratamiento antirretroviral, también denominado triterapia. El tratamiento antirretroviral está constituido por tres fármacos, dos de los cuales corresponden a los INTR, siendo de elección la 3a droga entre las familias de INNTR, IP, INSTI. Actualmente la tercera droga de elección en países desarrollado son los inhibidores de integrasa.
Los INTR son profármacos que poseen un mecanismo competitivo con los nucleósidos o nucleótidos fisiológicos, de los que difieren únicamente en pequeños cambios en la molécula de ribosa. Los INTR se incorporan a la cadena de DNA viral, interrumpiendo la elongación de la misma y como consecuencia, inhiben la replicación viral. De acuerdo a su estructura molecular se pueden dividir en análogos de bases púricas: adenosina (didanosina) y guanosina (abacavir) y análogos de bases pirimidínicas: timidina (zidovudina y estavudina) y citidina (emtricitabina, lamivudina). Estos fármacos requieren tres fosforilaciones en el interior de la célula para activarse. En cambio, tenofovir es un análogo de nucleótidos (análogo de adenina), por lo que requiere una fosforilación menos para activarse.
En el metabolismo de los INTR no interviene el sistema enzimático del citocromo P450, por tal motivo son poco susceptibles de generar interacciones metabólicas relevantes. ZDV y ABC se glucuronidan, por lo que otros fármacos que afecten la glucuronidación pueden modificar sus concentraciones. Sin embargo, las interacciones de los análogos de nucleósidos se deben fundamentalmente a la potenciación de su toxicidad, por ej. anemia con la asociación de AZT a ribavirina, cotrimoxazol, o ganciclovir, entre otros. Lamivudina, emtricitabina, estavudina y tenofovir se eliminan principalmente por vía renal. Se ha descrito aumento del riesgo de toxicidad renal al asociar tenofovir a algunos inhibidores de la proteasa potenciados con ritonavir. La combinación de tenofovir con otros fármacos nefrotóxicos debe evitarse en lo posible1–3.
Si bien los INTR poseen efectos adversos a corto plazo, hoy en día denominado “tolerancia”, los efectos adversos más característicos aparecen a largo plazo y se relacionan con su toxicidad mitocondrial4,5. Los mecanismos de toxicidad mitocondrial y celular son complejos, destacando entre otros el hecho de que estos fármacos, además de inhibir la transcriptasa reversa del virus, pueden inhibir la DNA polimerasa gamma mitocondrial. Aunque por su mecanismo de acción todos los análogos pueden producir toxicidad mitocondrial, se produce con más frecuencia con los análogos de timidina, fármacos actualmente en desuso y por lo tanto, en la práctica clínica estos efectos son infrecuentes o anecdóticos. Dependiendo del fármaco, las manifestaciones clínicas pueden ser muy variables: miopatía, neuropatía, esteatosis hepática y acidosis láctica, pancreatitis y lipoatrofia periférica (posiblemente esto ocurra todos los análogos, pero predominantemente con estavudina y zidovudina)6,7.
Actualmente se encuentren vigentes 6 fármacos, que por orden alfabético son: abacavir, didanosina, emtricitabina, lamivudina, tenofovir y zidovudina.
ABACAVIR (ABC)Corresponde a un análogo de purina, derivado carbocíclico de la desoxiguanosina. Este fármaco presenta un mecanismo de fosforilación enzimática único, por lo que es poco probable que compita con la fosforilación de otros análogos, debe transformarse en carbovir trifosfato, el cual es el metabolito activo.
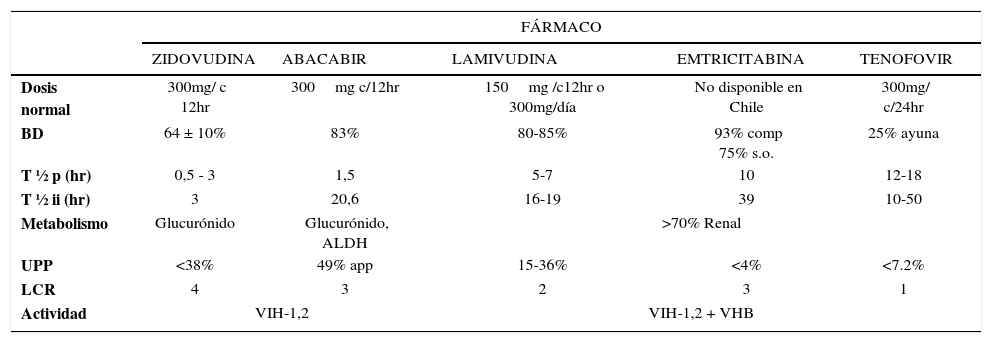
Su biodisponibilidad por vía oral es del 83% y presenta una buena difusión a tejidos, concentrándose en altas concentración en líquido céfalo-raquídeo (LCR) (alrededor del 30- 40%). Su vía de metabolización es a través de la glucuronidación y a través de la enzima alcohol deshidrogenasa. Pese a ello, su interacción con el etanol no se considera relevante. El resto de sus características farmacocinéticas se indican en la Tabla 1. Puede tomarse con o sin alimentos. No requiere ajuste de dosis en insuficiencia renal (Tabla 2).
{(TABLA 1)}] CARACTERÍSTICAS FARMACOCINÉTICAS INTR76
| FÁRMACO | |||||
|---|---|---|---|---|---|
| ZIDOVUDINA | ABACABIR | LAMIVUDINA | EMTRICITABINA | TENOFOVIR | |
| Dosis normal | 300mg/ c 12hr | 300mg c/12hr | 150mg /c12hr o 300mg/día | No disponible en Chile | 300mg/ c/24hr |
| BD | 64 ± 10% | 83% | 80-85% | 93% comp 75% s.o. | 25% ayuna |
| T ½ p (hr) | 0,5 - 3 | 1,5 | 5-7 | 10 | 12-18 |
| T ½ ii (hr) | 3 | 20,6 | 16-19 | 39 | 10-50 |
| Metabolismo | Glucurónido | Glucurónido, ALDH | >70% Renal | ||
| UPP | <38% | 49% app | 15-36% | <4% | <7.2% |
| LCR | 4 | 3 | 2 | 3 | 1 |
| Actividad | VIH-1,2 | VIH-1,2 + VHB | |||
BD: biodisponibilidad, T½p: vida media plasmática, T½ii: vida media intracelular, UPP: Unión a proteínas plasmáticas, LCR: penetración a líquido céfalo raquídeo.
Ref: Tabla confeccionada con datos extraídos de Micromedex® Healthcare Series y Lexi-comp®.
{(TABLA 2)}] AJUSTE POR DISFUNCIÓN RENAL Y ADMINISTRACIÓN POR SONDA NASOGÁSTRICA
| FÁRMACO | DOSIS HABITUAL | DOSIS EN IR INSUFICIENCIA RENAL | DOSIS EN HD/DP | SNG | |
|---|---|---|---|---|---|
| INTR | Abacavir (ABC) | 300mg/12h | No requiere ajuste de dosis. | Dosis habitual. HD: administrar independientemente de la sesión de HD, ya que se elimina mínimamente. | SI |
| Emtricitabina (FTC) | 200mg c/24h *Solo disponible asociado a TDF | Truvada©: no administrar a pacientes con Cl <30mL/min | No se ha estudiado en diálisis peritoneal. Truvada®: no administrar a pacientes en HD (administrar los componentes por separado, ajustando dosis adecuadamente) | SI | |
| Zidovudina (AZT) | 300mg/12h | Cl 10-50: 250-300mg c/12h. Cl <10: 300mg c/24h. En co-formulados: <50 mL/min (administrar los componentes por separado) | 300mg c/24h. HD/CAPD: no afecta la eliminación de AZT y aumenta la eliminación de GAZT. Por precaución, se recomienda administrar la dosis diaria postHD/CAPD | SI | |
| Tenofovir (TDF) | 300mg/24h | Cl ≥ 50: no requiere ajuste de dosis Cl 30-49: 300mg c/48h Cl 10-29: 300mg c/72 a 96h Cl > 10: no recomendado | HD: habitualmente 300mg una vez por semana, después de una de las sesiones (asumiendo 3 sesiones de diálisis semanales de 4h) | SI | |
| Lamivudina (3TC) | 150mg c/12h ó 300mg c/24h | Cl >50: 150mg c/12h ó 300mg c/24h Cl 30-49: 150mg c/24h Cl 15-29: 100mg c/24h Cl 5-14: 50mg c/24h Cl <5: 25mg c/24h | HD: 25mg c/24h (primera dosis 50 mg) Los días de la HD, administrar post-HD. | ||
| INNTR | Efavirenz (EFV) | 600mg/24h | No requiere ajuste de dosis. | SI | |
| Nevirapina (NVP) | 200mg/12h | No requiere ajuste de dosis. | HD: dosis Post HD o un suplemento de 200mg | ||
| Etravirina (ETR) | 200mg/12h o 400mg/24h | Sin datos de ajustes disponible. | SI | ||
| Rilpivirina (RPV) | 25mg/24h | IR leve-moderada: no requiere ajuste de dosis. IR grave: no hay datos. | SI | ||
| IP | Atazanavir (ATV) | 300mg/24h (+100mg/24h RTV) 400mg/24h | No requiere ajuste de dosis HD/CAPD: por su elevada unión a proteínas plasmáticas, no es de esperar que se elimine en las sesiones de HD/CAPD. | SI | |
| Darunavir (DRV) | 800mg/24h (+100mg/24h RTV) 600mg/12hrs (+100mg/12hrs RTV) | IR leve, moderada o grave: no requiere ajuste de dosis HD/CAPD: debido a la elevada unión a proteínas plasmáticas, no es de esperar que se elimine en las sesiones de HD/CAPD | SI | ||
| Lopinavir (LPV/r) | 400/100mg/12h | No requiere ajuste de dosis | SI | ||
| Ritonavir (RTV) | 100mg c/12hrs o 24/horas | Booster farmacológico no requiere ajuste | NO | ||
| INSTI | Raltegravir (RAL) | 400mg/12h | No requiere ajuste de dosis | SI | |
| Elvitegravir (EVG)/ cobicistat / TDF/FTC | 150/150/200/245mg | Cobicistat reduce levemente el filtrado glomerular renal estimado de creatinina (aunque no altera el filtrado glomerular real), debido a la inhibición de la secreción tubular de creatinina. La combinación EVG/COBI/TDF/FTC no debe iniciarse en pacientes con Cl<30mL/min | La combinación EVG/ COBI7TDF/FTC no debe emplearse si Cl <70ml/min | NO | |
| Dolutegravir (DTG) | 50mg/24h | No se requiere ajuste de dosis. | Sin datos | ||
| CCR5 | Maraviroc (MVR) | Dosis variable en función de los antirretrovirales asociados | En ausencia de inhibidores potentes del CYP3A4 no requiere ajuste de dosis. Sólo se recomienda un ajuste de dosis en pacientes con Cl <80ml/min, en estos casos administrar 150mg c/24h | HD: en ausencia de inhibidores potentes del CYP3A4 no se requiere ajuste de dosis. En presencia de los mismos, dosificar igual que para Cl | SI |
Ref: Datos extraídos de Micromedex® Healthcare Series y Lexi-comp
HD: Hemodialisis.
PD: Peritoneodiálisis.
Abacavir es un fármaco generalmente bien tolerado8, los efectos adversos más frecuentes son náuseas, dolor abdominal, malestar general y cefaleas, descritos en alrededor del 7% de los pacientes que han participado en los estudios de desarrollo clínico. Su reacción adversa más característica es la reacción de hipersensibilidad que ocurre en el 3-5% de los pacientes, existiendo una clara predisposición genética para la hipersensibilidad a dicho fármaco asociada principalmente al haplotipo HLA-B*5701, presentándose en más de la mitad de pacientes con este alelo y siendo absolutamente excepcional sin el mismo. Actualmente debe determinarse el HLA B*5701 a todos los pacientes antes de comenzar tratamiento con abacavir9. Si el resultado es positivo, no se debe utilizar ABC. En caso de sospecha de reacción de hipersensibilidad a abacavir no debe reintroducirse jamás el fármaco por la posibilidad de presentar una reacción severa, la cual suele manifestarse como un síndrome multiorgánico con una erupción cutánea eritematosa, asociada a síntomas más inespecíficos como fiebre, náuseas, vómitos, malestar, diarrea, mialgias o artralgias. Por otro lado, algunos estudios han sugerido que abacavir incrementa el riesgo cardiovascular, pero dicha evidencia sigue aun siendo controversial10.
Emtricitabina (FTC)Es análogo fluorado de citosina, con actividad frente al virus de la hepatitis B. Su biodisponibilidad varía de acuerdo a su forma farmacéutica, siendo un 93% para su presentación en cápsulas y de un 75% para la solución oral. Puede administrarse con o sin alimentos. Se elimina mayoritariamente por vía renal mediante filtración glomerular y secreción tubular activa (86% inalterado) y se recomienda ajustar la dosis en caso de insuficiencia renal, por lo que no deben usarse coformulados en pacientes con un clearance de creatinina menor de 50ml/min11,12. (Tabla 2). El resto de sus características farmacocinéticas se indican en la Tabla 1.
Es un fármaco muy bien tolerado y no suele asociarse a efectos adversos serios12, emtricitabina no se comercializa individualmente en Chile, no obstante es posible encontrarlo en asociación con otros ARV (Truvada® comprimidos con emtricitabina 200mg y tenofovir 245mg y Atripla® comprimidos que contienen emtricitabina 20mg, tenofovir 245mg y efavirenz 600mg).
Lamivudina (3TC)Es análogo de la citosina, con actividad frente al virus de la hepatitis B, siendo muy bien tolerado y difícilmente puede atribuírsele efectos adversos graves. Sin embargo, existen reportes en la actualidad de reacciones de hipersensibilidad asociadas13.
Presenta una biodisponibilidad oral del 86% y no se modifica con los alimentos. Difunde escasamente a través de la barrera hematoencefálica y se elimina principalmente por la orina (5-10% por metabolismo hepático). Lamivudina se elimina principalmente por vía renal (70% inalterado) y su dosis debe ajustarse en pacientes con insuficiencia renal (Tabla 2). El resto de sus características farmacocinéticas se indican en la Tabla 1.
La barrera genética del fármaco es muy baja y la mayoría de pacientes en los que se produce un fallo virológico con una combinación que incluye lamivudina desarrollan la mutación M184V/I que confiere resistencia completa a la misma14.
Tenofovir Desoxi Fumarato (TDF)Tenofovir es actualmente el INTR más ampliamente utilizado en la práctica clínica, junto con emtricitabina o lamivudina15. Posee actividad frente a VIH y VHB, siendo fármaco de elección en pacientes co-infectados con VHB.
Se administra por vía oral como profármaco en forma de “disoproxil fumarato (DF)” y con alimentos para mejorar su biodisponibilidad (biodisponibilidad de 25% en ayunas y 40% con alimentos). Requiere ser bifosforilado dentro de la célula para ser una molécula activa. Su vida media intracelular es mayor de 30 horas, lo cual permite su administración en una sola toma diaria. La mayor parte del fármaco se excreta inalterado en orina (70-80%), tanto por filtración como por un sistema de transporte tubular activo, y la dosis debe ajustarse en caso de insuficiencia renal (Tabla 2). Los inhibidores de la proteasa potenciados aumentan ligeramente las concentraciones de tenofovir sin que sea preciso ajustar las dosis1–3. El resto de sus características farmacocinéticas se indican en la Tabla 1.
Es un fármaco generalmente muy bien tolerado, con un excelente perfil metabólico y sin potencial toxicidad mitocondrial. La toxicidad renal es su efecto adverso más característico, potenciándose si se coadministra con otros fármacos nefrotóxicos16. También se ha descrito con TDF una mayor pérdida de la densidad mineral ósea tanto en columna como en cadera, asociado a un mayor riesgo de fractura17,18.
Actualmente existe disponible una nueva formulación de tenofovir denominado tenofovir alafenamida o TAF, el mecanismo de acción de TAF no deja de ser el mismo que el de tenofovir disoproxil fumarato, ya que la parte de la molécula activa es la misma. No obstante, TAF presenta una especial afinidad por los linfocitos que hace que sus concentraciones intracelulares lleguen a ser cinco veces las observadas en sangre y permite dosis mucho más reducidas sin reducir su eficacia contra el virus, eso sí, con un menor impacto a nivel renal y óseo16–18.
Zidovudina (AZT)Fue el primer antiretroviral disponible para el tratamiento del VIH19. Se sintetizó en 1964 y se utilizó con poco éxito en el tratamiento de tumores. En 1987 fue aprobado por la FDA para el tratamiento de la infección por VIH. Su estructura química corresponde a un análogo de timidina, sufre trifosforilación en el citoplasma por la timidin-quiinasa celular y en su forma trifosfafatada actúa como inhibidor competitivo de timidina trifosfato.
Se puede administrar con o sin alimentos. Zidovudina es metabolizada principalmente mediante glucuronidación hepática, aunque el 25% se elimina inalterada por orina y se requiere ajuste de dosis en insuficiencia renal (Tabla 2)1–3. El resto de sus características farmacocinéticas se indican en la Tabla 1. Sus principales efectos adversos son la mielotoxicidad, especialmente la anemia, reversible tras la retirada del fármaco y la pérdida de grasa subcutánea (lipoatrofia), menos importante que con estavudina, pero también estigmatizante y muy difícilmente corregible. Mucho menos frecuentes son otras manifestaciones de toxicidad mitocondrial (miopatía, cardiopatía)20,21.
Durante mucho tiempo se utilizó ampliamente en la práctica clínica habitual, considerándose que jugaba un papel especial en situaciones clínicas tales como la trombocitopenia asociada al VIH, la profilaxis post-exposición y la prevención de la transmisión vertical. Actualmente su uso, en países desarrollados o de altos ingresos ha sido discontinuado frente a la disponibilidad de INTR menos tóxicos6.
INHIBIDORES NO NUCLEOSÍDICOS DE LA TRANSCRIPTASA REVERSA (INNTR)A diferencia de los INTR, estos se caracterizan por ser drogas activas, actuar a través de un mecanismo no competitivo y en términos moleculares, se unen directamente y de manera reversible al centro catalítico de la transcriptasa reversa o en un sitio cercano al mismo, provocando cambios conformacionales en la enzima que inhibe la DNA polimerasa, tanto la dependiente de DNA como RNA. Son fármacos activos específicos sobre el VIH-1, sin que ser activos frente a cepas del VIH-1 del grupo O, ni frente al VIH-2, ni frente a retrovirus animales5.
Poseen un metabolismo predominantemente hepático, en este intervienen diferentes isoenzimas del citocromo P450, especialmente de CIP3A4, y también por glucuronoconjugación (Tabla 3)1–3. Se caracterizan por ser potentes inductores de CIP3A4 y de otras enzimas, pudiendo tener la capacidad de generar interacciones metabólicas muy relevantes, que pueden llevar a la pérdida de efectividad de otros tratamientos. Sin embargo, en algunos casos pueden comportarse además como inhibidores enzimáticos, dando lugar a interacciones complejas, con el riesgo de generar problemas de seguridad asociado a dichas interacciones. Debido a su efecto inductor como se indicaba en el párrafo anterior, los INNTR pueden dar lugar a una reducción de la efectividad de muchos fármacos, tales como anticonceptivos orales, estatinas o antimicrobianos como claritromicina, itraconazol o ketoconazol. En el caso de efavirenz, la competencia por la metabolización vía CIP3A4 puede producir inhibición del metabolismo y con ello, la aparición de efectos adversos graves y/o potencialmente mortales con algunos fármacos tales como: terfenadina, cisaprida, midazolam, triazolam, pimozida, bepridil o alcaloides ergóticos, por lo que no deben emplearse en combinación. Efavirenz y etravirina también pueden comportarse como inhibidores de las isoenzimas 2C9/19 y es por ello que podrían aumentar el efecto de los fármacos que se eliminan por estas vías como el voriconazol y acenocumarol. Por otro lado, no solo los INNTR son capaces de modificar la respuesta de otros fármacos sino también vice-versa; fármacos tales como, anticonvulsivante de primera generación como carbamazepina, fenitoína e incluso algunos productos naturales como el Hypericum (o hierba de San Juan), pueden reducir la eficacia de este grupo de ARV1–3.
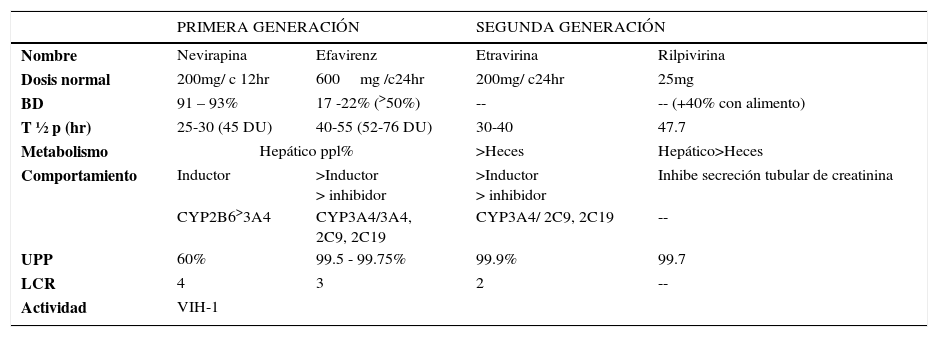
CARACTERÍSTICAS FARMACOCINÉTICAS INNTR
| PRIMERA GENERACIÓN | SEGUNDA GENERACIÓN | |||
|---|---|---|---|---|
| Nombre | Nevirapina | Efavirenz | Etravirina | Rilpivirina |
| Dosis normal | 200mg/ c 12hr | 600mg /c24hr | 200mg/ c24hr | 25mg |
| BD | 91 – 93% | 17 -22% (>50%) | -- | -- (+40% con alimento) |
| T ½ p (hr) | 25-30 (45 DU) | 40-55 (52-76 DU) | 30-40 | 47.7 |
| Metabolismo | Hepático ppl% | >Heces | Hepático>Heces | |
| Comportamiento | Inductor | >Inductor > inhibidor | >Inductor > inhibidor | Inhibe secreción tubular de creatinina |
| CYP2B6>3A4 | CYP3A4/3A4, 2C9, 2C19 | CYP3A4/ 2C9, 2C19 | -- | |
| UPP | 60% | 99.5 - 99.75% | 99.9% | 99.7 |
| LCR | 4 | 3 | 2 | -- |
| Actividad | VIH-1 | |||
BD: Biodisponibilidad, T½p: Vida media plasmática, UPP: Unión a proteínas plasmáticas, LCR: Penetración a líquido céfalo raquídeo, DU: Dosis única
Ref: Tabla confeccionada con datos extraídos de Micromedex® Healthcare Series y Lexi-comp®76.
Las reacciones de hipersensibilidad, especialmente en forma de exantema cutáneo, son los efectos adversos más frecuentemente asociados a esta familia, especialmente con nevirapina y etravirina. La hepatotoxicidad también aparece con cierta frecuencia particularmente con nevirapina, con efavirenz también existe riesgo de exantema durante las primeras semanas de tratamiento. Las alteraciones del sistema nervioso central (sueños vívidos, mareos, insomnio, depresión) son características propias de efavirenz22.
Actualmente se encuentran disponibles un total de 4 INNTR: dos de primera generación (efavirenz y nevirapina) y dos de segunda generación (etravirina y rilpivirina).
INHIBIDORES DE LA PROTEASA (IP)Los IP son drogas activa que no requieren ninguna transformación intracelular para su actual23. Inhiben la enzima encargada de la maduración de las proteínas virales e inhiben de forma potente la replicación viral. La inhibición de la proteasa no impide que se sinteticen los grandes polipéptidos virales codificados por gag y gag-pol, pero al no fragmentarse no son funcionales y no se producen virus con capacidad infectiva. Los IP tienen una estructura química parecida a los péptidos virales sustrato de la proteasa, con una elevada afinidad para el dominio activo de la misma, inhibiendo su actividad catalítica5.
Los primeros IP tenían grandes inconvenientes debido a sus reacciones adversas, por lograr concentraciones plasmáticas en el límite inferior del rango terapéutico y por una compleja posología que limitaban enormemente su eficacia terapéutica24. Con el descubrimiento del uso del ritonavir a dosis bajas como potenciador (booster) de los otros IP, se logró aumentar sus concentraciones plasmáticas por su efecto inhibitorio sobre el citocromo P450, cambiando de forma radical la eficacia de esta familia23,25. Sus características farmacocinéticas se describen en la Tabla 4.
CARACTERÍSTICAS FARMACOCINÉTICAS INHIBIDORES PROTEASA
| FÁRMACO | |||||
|---|---|---|---|---|---|
| LOPINAVIR/RIT | ATAZANAVIR | DARUNAVIR | FOSAMPRENAVIR | SAQUINAVIR | |
| Dosis normal | 400/100 c/12hr | 300 + 100RTV c/24hr 400 c/24hr | 300 + 100 RTV c/12hr | 700 +100 RTV c/12hr | 500 + 100RTV c/12hr |
| BD | --- | 68% | 37%; 82 DM | --- | 4% sin booster |
| T ½ p (hr) | 5 -6 | 12 (ATV/r); 6,5 (400) | 15 (DRV/r) | 15-23(FPV/r); 7,7(FPV) | 7 |
| Metabolismo | Hepático | ||||
| Comportamiento | >Inhibidor > Inductor | Inhibidor | Inhibidor débil | ||
| 3A4; 2D6/ 3A4, 2C9, 2C19 | 3A4; UDPGT 1A1 | 3A4 | 3A4 | 3A4 | |
| UPP | 98-99% | 86% | 95% | 90% | 97% |
| LCR | 3 | 2 | 3 | 2 | 1 |
| Actividad | VIH-1,2 | VIH-1 | VIH-1,2 | VIH-1,2 | VIH-1,2 |
BBD: Biodisponibilidad, T½p: Vida media plasmática, UPP: Unión a proteínas plasmáticas, LCR: Penetración a líquido céfalo raquídeo, DU: Dosis única
Ref: Tabla confeccionada con datos extraídos de Micromedex® Healthcare Series y Lexi-comp®76.
Todos los IP se metabolizan por vía hepática (isoenzimas del citocromo P450). Las principales interacciones medicamentosas relacionadas con los IP ocurren como, en gran medida, como resultado de la inducción o inhibición del CIP3A4, Por ello, ninguno de ellos necesita ajuste de dosis en pacientes con insuficiencia renal crónica, por lo que no hay recomendaciones específicas en ese sentido (Tabla 2)1–3. En la mayor parte de los casos se utilizan siempre potenciados con ritonavir, comportándose como potentes inhibidores de CIP3A4 y de otras isoenzimas y proteínas transportadoras. Debido a su efecto inhibitorio se producen interacciones farmacocinéticas relevantes con fármacos tales como inmunodepresores (ciclosporina, tacrolimus, sirolimus), estatinas o antagonistas del calcio, entre otros. Por este motivo, no se recomienda asociar IP boosteados por el riesgo de toxicidad cuando se administran con otras drogas que comparten la misma ruta metabólica vía CIP450: amiodarona, analgésicos opiáceos (dextropropoxifeno, meperidina), benzodiacepinas (diazepam, flurazepam, midazolam [por vía oral], triazolam)26–29, clozapina, colchicina (en caso de insuficiencia renal o hepática), derivados de la ergotamina, drogas de abuso (éxtasis, metanfetamina), estatinas (lovastatina, simvastatina y atorvastatina en dosis mayores a 20mg/dia)30, inhibidores de la 5-fosfodiesteresas como sildenafil (pueden emplearse dosis reducidas para la disfunción eréctil, pero su uso está contraindicado en la hipertensión pulmonar, en el caso particular de dicho fármaco31,32. Su uso además está contraindicado con formas farmacéuticas de administración no-oral como algunos fármacos inhalados como fluticasona, budesonida33–36 (síndrome de Cushing) o salmeterol (prolongación del intervalo QT)37,38.
Por otro lado, ritonavir también tiene un efecto inductor de varias isoenzimas del citocromo P450 como el CYP1A2 o 2C9/19, así como de la glucuronidación. Por este motivo puede reducir la eficacia de algunos fármacos que se eliminan por estas vías como el voriconazol y acenocumarol39. Al igual que nos INNTR, varios son los fármacos que pueden afectar la efectividad de estos ARV; por ejemplo, en pacientes bajo tratamiento antituberculoso con rifampicina/rifabutina, no deben utilizarse IP por la disminución de sus niveles plasmáticos y mayor riesgo de falla virológica40. Esta disminución se observa también con el uso de la hierba de San Juan por el mismo motivo. Una interacción relevante en pacientes hospitalizados son las reacciones de tipo Antabus® cuando se asocian a disulfiram, metronidazol o sulfonilureas a formulaciones líquidas de estos ARVs por su elevado contenido de etanol (RTV con 43% v/v y LPV/r con 42% p/p). A diferencia de la solución oral, ni las cápsulas ni los comprimidos de Kaletra® (LPV/r) contienen etanol41.
Una característica importante de los IP potenciados es su elevada barrera genética para el desarrollo de resistencias, debiendo acumularse múltiples mutaciones para que el virus sea resistente. Cuando ya existen mutaciones de resistencia en la proteasa la barrera genética se reduce y en los fallos virológicos pueden aparecer nuevas mutaciones. En esta situación se ha observado que los IP de última generación, especialmente DRV, ofrecen una barrera genética más elevada que los otros IP5,23.
Algunos efectos adversos de los IP se han considerado de familia, especialmente las alteraciones gastroinstestinales (diarrea, náuseas, vómitos, dolor abdominal) y metabólicas (dislipidemia, resistencia a la insulina, diabetes mellitus)23. La dislipidemia aparece hasta en el 70% de los pacientes en tratamiento con IP y normalmente requiere de la instauración de tratamiento hipolipemiante. A pesar de ello, existen diferencias notables entre los diferentes IP, los que con mayor frecuencia se asocian con diarrea y otras alteraciones digestivas de intensidad moderada a grave son lopinavir/ritonavir (LPV/r) y fosamprenavir/r (FPV/r), mientras que con atazanavir/r (ATV/r) o darunavir/r (DRV/r) la incidencia es considerablemente menor. Asimismo, los IP con un mejor perfil metabólico son ATV/r y DRV/r.
Para el caso particular de atazanavir, la hiperbilirrubinemia indirecta es común, debido a la inhibición de la enzima glucuronil transferasa por efecto del ARV, generando mayor cantidad de bilirrubina no conjugada en circulación. Generalmente no requiere la interrupción del tratamiento en ausencia de elevación concomitante de los las transaminasas hepáticas. Nefrolitiasis por depósito tubular de cristales: menos común con ATV y DRV, anomalías de conducción cardíaca: (bloqueo aurículo-ventricular) se desarrollan en el 5% de los pacientes que recibieron ATV y se han comunicado con otros IP (RTV, LPV/r).
Actualmente están comercializados en Chile 5 IP, que por orden alfabético son: ATV, DRV, FPV, LPV y SQV. Como se señalaba anteriormente RTV no se utiliza como antiretroviral, sino como booster o potenciador farmacológico a bajas dosis. Últimamente se incorporó otro potenciador o booster al mercado, el cobicistat el cual es un potenciador selectivo del citocromo P450 de las isoenzimas CIP3A4, y es un débil inhibidor del CIP2D6, no posee de inducción enzimatica en comparación a ritonavir, y su impacto en el perfil lipídico es levemente mejor que ritonavir. Se encuentra asociado bajo el nombre de STRIBILD®5,78.
INHIBIDORES DE LA FUSIÓNEn esta familia existe solo un representante: enfuvirtide (T-20), el cual es capaz de inhibir la fusión de la cubierta viral del VIH con la membrana celular, impidiendo el ingreso del contenido viral en los linfocitos. Su actividad frente al VIH es independiente de correceptor utilizado (CCR5 y/o X4) y su perfil de resistencias es completamente diferente al de los otros ARV. Su uso en la actualidad está reservado para pacientes con virus multirresistentes. La barrera genética de T-20 es baja y si no se acompaña de otros fármacos activos rápidamente aparecen mutaciones de resistencia, perdiendo su eficacia5,41.
T-20 se comercializa como Fuzeon® en jeringas de 90mg y debe administrarse por vía subcutánea. La dosificación en adultos es de 90mg cada 12hr y el paciente debe recibir una capacitación para su autoadministración. Su efecto adverso más característico es la aparición de máculas dolorosas, por lo cual se debe rotar el sitio de inyección42.
La biodisponibilidad tras su administración subcutánea es del 80%. Se elimina mediante catabolismo de sus aminoácidos constituyentes y no es sustrato ni influye en la actividad de ninguno de los sistemas metabólicos de los otros ARV, por lo que no es susceptible de presentar interacciones metabólicas relevantes. No se requiere ajuste de dosis en insuficiencia hepática ni renal. El resto de sus características farmacocinéticas se indican en la Tabla 5.
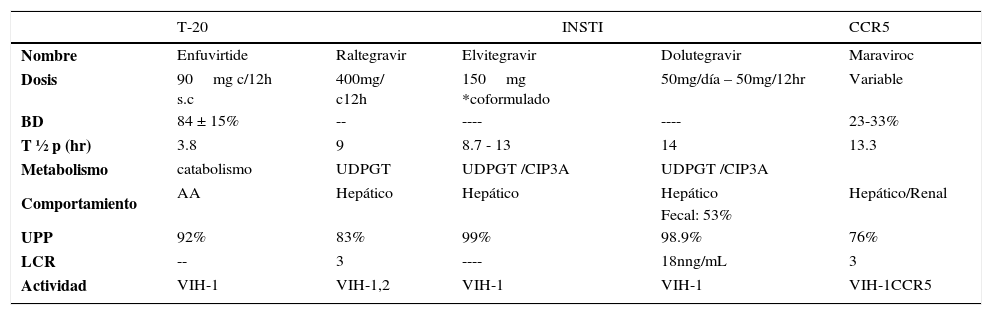
CARACTERÍSTICAS FARMACOCINÉTICAS OTRAS FAMILIAS
| T-20 | INSTI | CCR5 | |||
|---|---|---|---|---|---|
| Nombre | Enfuvirtide | Raltegravir | Elvitegravir | Dolutegravir | Maraviroc |
| Dosis | 90mg c/12h s.c | 400mg/ c12h | 150mg *coformulado | 50mg/día – 50mg/12hr | Variable |
| BD | 84 ± 15% | -- | ---- | ---- | 23-33% |
| T ½ p (hr) | 3.8 | 9 | 8.7 - 13 | 14 | 13.3 |
| Metabolismo | catabolismo | UDPGT | UDPGT /CIP3A | UDPGT /CIP3A | |
| Comportamiento | AA | Hepático | Hepático | Hepático | Hepático/Renal |
| Fecal: 53% | |||||
| UPP | 92% | 83% | 99% | 98.9% | 76% |
| LCR | -- | 3 | ---- | 18nng/mL | 3 |
| Actividad | VIH-1 | VIH-1,2 | VIH-1 | VIH-1 | VIH-1CCR5 |
BD: Biodisponibilidad, T1/2: Vida media plasmática, T1/”ii: Vida media intracelular, UPP: Unión a proteínas plasmáticas, LCR: Penetración a líquido céfalo raquídeo, UDPGT: Uridinadifosfato glucuroniltransferasa.
Ref: Tabla confeccionada con datos extraídos de Micromedex® Healthcare Series y Lexi-comp®76.
Solo disponemos de un inhibidor de los correceptores CCR5: maraviroc. MVC está indicado en el tratamiento de la infección por el VIH-1 con tropismo CCR5 detectable mediante test tropismo validado y en combinación con otros fármacos ARV. Actúa bloqueando la entrada del VIH a las células humanas. No es eficaz frente a virus con tropismo dual, mixto o X45,43.
MVC se comercializa como Celsentri® en comprimidos de 150 y 300mg. La dosis indicada en el adulto es de 150mg, 300mg o 600mg dos veces al día, dependiendo de las interacciones con la terapia antirretroviral y con otros medicamentos.
Sus características farmacocinéticas se indican en la Tabla 5. MVC es sustrato de CYP3A4, pero no es inhibidor ni inductor del mismo. Los inhibidores e inductores de CYP3A4 alteran profundamente los parámetros farmacocinéticos de MVC, recomendándose cambios en su dosis, sin que MVC modifique significativamente las concentraciones de otros fármacos. En general se ajustarán las dosis como sigue: 150mg/12h cuando se administra con inhibidores del CYP3A4 como por ejemplo IP/r (con excepción de TPV/r y FPV/r); 600mg BID cuando se administra con fármacos inductores como EFV o rifampicina (con excepción de NVP), en ausencia de inhibidores potentes, en cuya presencia predomina el efecto inhibidor y se administrarán 150mg BID; 300mg BID con otros fármacos (incluyendo TPV/r, FPV/r y NVP). MVC requiere ajuste de dosis en insuficiencia renal en caso de asociarse a inhibidores potentes del CYP3A4 y se han descrito aumentos de sus concentraciones plasmáticas en pacientes con insuficiencia hepática, aunque se desconoce la posible importancia clínica que ello pueda suponer5.
El perfil de resistencias de MVC es completamente diferente al de los otros ARV. Hasta el momento se han descrito dos mecanismos principales de resistencia. El primero y más frecuente es la emergencia de virus con tropismo X4 preexistentes como población minoritaria al inicio del tratamiento. El segundo mecanismo resulta de la selección de mutaciones en diferentes regiones de la envoltura (gp 120) que impiden la interacción entre el virus y el receptor CCR544.
Es un fármaco habitualmente muy bien tolerado, con un excelente perfil metabólico y de tolerabilidad digestiva y sin efectos adversos particulares43.
INHIBIDORES DE LA INTEGRASA (INSTI)Los inhibidores de la integrasa actúan en un paso replicativo del VIH diferente del resto de las familias de ARV, bloqueando el paso de transferencia de hebra del proceso de integración45,46. El sitio activo de la integrasa se une al ADN de la célula del huésped e incluye 2 cationes de metal divalentes, que sirven como objetivos de quelación para los INSTI. Como resultado, cuando está presente el INSTI, el sitio activo de la enzima es ocupado y el proceso de integración se detiene. Tal como es el caso de los INNTR, tenemos fármacos de primera generación (Raltegravir y Elvitegravir) y de segunda generación (dolutegravir). DTG presenta algunas diferencias con los otros 2 INSTI aprobados, que le confieren una mayor barrera genética. Sus características farmacocinéticas se detallan en la Tabla 5. Actualmente hay un nuevo fármaco en estudio Cabotegravir (CBT).
Raltegravir (RAL) presenta una baja barrera genética con alta resistencia cruzada con otros INI. La Resistencia se presenta por mutaciones N155H/S,Q148H/K/R y por la Y143R/H/C,E92Q la L74M,E138A/K y G140S, no es sustrato ni influye en la actividad del citocromo P450. Se metaboliza por glucuronidación (UGT1A1), sin inhibir ni inducir esta enzima. Los inhibidores e inductores de UGT1A1 modifican los parámetros farmacocinéticos de RAL, pero en la mayoría de los casos no se recomiendan cambios en su dosificación por el amplio margen terapéutico de este fármaco47. El año 2015 se aprobó tanto por parte de la FDA como de la EMA Dutrebis®, que es una coformulación a dosis fijas de RAL 300mg más 3TC 150mg para su uso en combinación siempre con otros antirretrovirales. No se trata de una biterapia. Uno de los beneficios que aportaría este combo es una nueva formulación de RAL que permite dosis más bajas.
Elvitegravir (EVG) se metaboliza principalmente a través del CYP3A4. Está contraindicada la administración concomitante de EVG con inductores potentes del CYP3A4, tales como la Hierba de San Juan y fármacos como: rifampicina, carbamazepina, fenobarbital y fenitoína). Su biodisponibilidad varía según la alimentación, siendo recomendado su ingesta con una comida de alto contenido graso (en al menos 500Kcal, 50% de grasa). En general es un fármaco bien tolerado, presentando reacciones adversas fundamentalmente de tipo gastrointestinal como náuseas o diarrea. Las diarreas de hecho son el efecto secundario que aparece con mayor frecuencia, con un porcentaje del 25%. Otros efectos secundarios que aparecen con alguna frecuencia con cefalea y reacciones cutáneas. También habría que destacar los aumentos de los niveles de creatinina sérica, aunque es un efecto imputable al potenciador cobicistat. EVG es un nuevo inhibidor de la integrasa cuya principal ventaja frente a RAL radica en su potencial para ser coformulado en una sola pastilla. Las limitaciones incluyen requisitos de ajuste de dosis, bastantes interacciones y una barrera genética a la resistencia relativamente baja79.
Dolutegravir (DTG) es actualmente la tercera droga recomendada a nivel internacional como de primera línea, dado su administración en una toma diaria, bajo perfil de toxicidad e interacciones, además de contar con una barrera genética alta45,46. La dosis recomendada de dolutegravir es de 50mg (un comprimido) por vía oral una vez al día para pacientes sin resistencia documentada. Si esta resistencia está documentada, la dosis debe aumentar a 50mg dos veces al día. Puede tomarse con o sin alimentos, aunque en pacientes con resistencia a inhibidores de la integrasa (sobre todo para mutación Q148) se recomienda administrarlo con alimentos. Es metabolizado por UGT1A1 y en menor grado por CYP3A4, sin que sea inductor ni inhibidor de los sistemas metabólicos habituales. Su potencial de interacciones es muy escaso y puede administrarse a las dosis habituales con la mayoría de fármacos. La difusión de DTG al líquido cefalorraquídeo es buena y también se alcanzan concentraciones eficaces en tracto genital femenino y masculino. En cuanto a sus concentraciones en LCR, se ha postulado que DTG podría alcanzar concentraciones en el sistema nervioso central capaces de inhibir la reproducción del VIH. Este hecho resulta muy conveniente, ya que podría significar que el fármaco sería capaz de reducir el tamaño de los reservorios y minimizar la replicación residual del virus que se produce incluso durante un tratamiento antirretroviral eficaz79.
No es preciso ajustar la dosis de DTG ni en pacientes con insuficiencia renal ni con insuficiencia hepática leve o moderada. Los únicos fármacos con los que se recomienda aumentar la dosis de dolutegravir (50mg/12h) son efavirenz, nevirapina, fosamprenavir/r, rifampicina, carbamazepina, fenitoína y fenobarbital. No se recomienda coadministrar dolutegravir con etravirina sin un inhibidor de la proteasa, ni con hierba de San Juan. DTG debe administrarse 2 horas antes o 6 horas después de antiácidos o productos con cationes polivalentes. DTG puede disminuir la secreción tubular renal de sustancias que se excretan vía OCT2, con un ligero incremento inicial de creatinina, sin que ello suponga toxicidad renal. También podría aumentar las concentraciones de metformina recomendándose vigilancia por si se requiere ajuste de dosis. En definitiva, dolutegravir tiene un excelente perfil farmacocinético y de interacciones en comparación al resto de las familias y con otros fármacos48,50.
Los INSTI son una familia de fármaco habitualmente muy bien tolerado, con un excelente perfil metabólico y de tolerabilidad digestiva. En general es un fármaco bien tolerado. Las reacciones adversas observadas con más frecuencia durante el tratamiento fueron náuseas (15%), diarrea (16%) y cefalea (14%)47. La reacción adversa más grave, vista en un único paciente, fue una reacción de hipersensibilidad que incluyó erupción y efectos hepáticos graves. Durante las primeras semanas se observaron ligeros aumentos en la creatinina sérica, no obstante no se consideraron clínicamente relevantes. Recientemente se ha documentado hasta un 16% de discontinuación por efectos adversos en una cohorte de pacientes en la vida real, aproximadamente un tercio de los pacientes que interrumpieron reportaron alteraciones del sueño (31,3%) y una proporción similar (29,5%) experimentaron problemas gastrointestinales, problemas neuropsiquiátricos fueron reportados por el 19,7% de los pacientes que interrumpieron dolutegravir81.
Cabotegravir (CTG): También conocido como GSK1265744 o GSK744 es un inhibidor de la integrasa, análogo de dolutegravir, actualmente se encuentra adporta de su comercialización. Ha mostrado actividad prolongada frente una amplia gama de cepas del VIH, con una elevada barrera genética. En la actualidad se estudian dos formas de cabotegravir, tabletas de administración oral (también conocidas como cabotegravir o CAB oral (10, 30, 60mg) y una forma inyectable de acción prolongada para aplicación intramuscular (conocida como cabotegravir-LA o CAB-LA) se administra bimensual 600mg y 800mg trimensual. El uso de este tipo de medicamento podría significar menos frecuencia, lo cual simplificaría la administración del régimen de tratamiento cada 2 o 3 meses80,82.
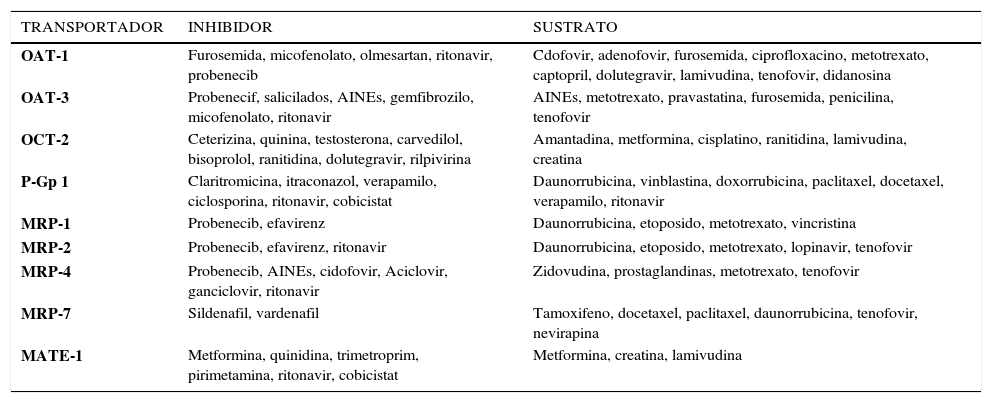
FUNCIÓN RENAL Y ARVSUn aspecto relevante de la terapia antiretroviral es la evaluación de la función renal en este grupo de pacientes. Su importancia radica en la creciente información científica sobre el envejecimiento y las comorbilidades asociadas. La exposición a nefrotóxicos es frecuente, y varios fármacos antirretrovirales que se utilizan en la actualidad poseen un potencial nefrotóxico. Por otro lado, algunos ARVs inhiben a nivel renal determinados transportadores tubulares que incrementan los valores de creatinina sérica, lo cual altera las estimaciones en la tasa de filtración glomerular, sin observarse necesariamente cambios en la tasa real de ésta51–53. Además, algunos antirretrovirales, tales como: TDF, ATV y LPV se han asociado con falla renal aguda, enfermedad renal crónica o progresión de la ERC, disfunción renal tubular, síndrome de Fanconi o a formación de cálculos renales54–58 (Tabla 6). Los mecanismos por los que estos fármacos antirretrovirales causan toxicidad renal son complejas y no del todo comprendidas actualmente (Tabla 6). Otros fármacos, tales como ritonavir (booster farmacológico), cobicistat (booster farmacológico), dolutegravir y rilpivirina pueden inhibir transportadores de fármacos que disminuyen la secreción tubular de creatinina59–63. En la Tabla 7 se describen los mecanismos de transporte tubulares, sus sustratos e inhibidores.
EFECTO DE LOS ARV SOBRE LA FUNCIÓN RENAL
| FAMILIA | EFECTO |
|---|---|
| INRTI | TDF: Falla renal aguda, aumento del clearance de creatinina, disfunción tubular proximal, proteinuria, hipofosfatemia, hipokalemia, síndrome de fanconi, acidosis metabólica |
| INNRTI | RPV: inhiben la secreción tubular de creatinina |
| IP | ATV y LPV/r: Incrementar el riesgo de progresión a ERC ATV: reportes de nefrolitiasis, falla renal aguda y nefritis intersticial aguda DRV: reportes de cristaluria y nefrolitiasis |
| INSTI | Elvitegravir/cobi, DTG: inhiben la secreción tubular de creatinina sin afectar la filtración glomerular |
ERC: Enfermedad renal crónica, TDF: Tenofovir, RPV: Tilpivirina, ATV: Atazanavir, LPV/r: Lopinavir/ritonavir, DRV: Darunavir, DTG: Dolutegravir.
Ref: Tabla confeccionada con datos extraídos de Micromedex® Healthcare Series y Lexi-comp®76.
TRANSPORTADORES TUBULARES RENALES, SUSTRATOS E INHIBIDORES
| TRANSPORTADOR | INHIBIDOR | SUSTRATO |
|---|---|---|
| OAT-1 | Furosemida, micofenolato, olmesartan, ritonavir, probenecib | Cdofovir, adenofovir, furosemida, ciprofloxacino, metotrexato, captopril, dolutegravir, lamivudina, tenofovir, didanosina |
| OAT-3 | Probenecif, salicilados, AINEs, gemfibrozilo, micofenolato, ritonavir | AINEs, metotrexato, pravastatina, furosemida, penicilina, tenofovir |
| OCT-2 | Ceterizina, quinina, testosterona, carvedilol, bisoprolol, ranitidina, dolutegravir, rilpivirina | Amantadina, metformina, cisplatino, ranitidina, lamivudina, creatina |
| P-Gp 1 | Claritromicina, itraconazol, verapamilo, ciclosporina, ritonavir, cobicistat | Daunorrubicina, vinblastina, doxorrubicina, paclitaxel, docetaxel, verapamilo, ritonavir |
| MRP-1 | Probenecib, efavirenz | Daunorrubicina, etoposido, metotrexato, vincristina |
| MRP-2 | Probenecib, efavirenz, ritonavir | Daunorrubicina, etoposido, metotrexato, lopinavir, tenofovir |
| MRP-4 | Probenecib, AINEs, cidofovir, Aciclovir, ganciclovir, ritonavir | Zidovudina, prostaglandinas, metotrexato, tenofovir |
| MRP-7 | Sildenafil, vardenafil | Tamoxifeno, docetaxel, paclitaxel, daunorrubicina, tenofovir, nevirapina |
| MATE-1 | Metformina, quinidina, trimetroprim, pirimetamina, ritonavir, cobicistat | Metformina, creatina, lamivudina |
OAT: Transportador de aniones orgánicos, OCT: Transportador de cationes orgánicos, P-Gp: Glicoproteína-P
Ref: Traducido de Jean C. Yombia, et al. AIDS 2014, 28:621–63277.
El daño hepático relacionado con los antiretrovirales es una causa frecuente de morbilidad, mortalidad y de interrupción del tratamiento64. Prácticamente todos los medicamentos antirretrovirales se han asociado con elevación de las enzimas hepáticas, aunque ciertos medicamentos pueden causar daño hepático con mayor frecuencia que otros. Además, ciertas comorbilidades, tales como la hepatitis B (VHB) o hepatitis C (VHC), pueden predisponer a los pacientes a esta situación65. Se han descrito 4 mecanismos principales de daño hepático por fármacos: incluidos los daños metabólicos mediados por el huésped, las reacciones de hipersensibilidad, la toxicidad mitocondrial, y los fenómenos de reconstitución inmune.
La injuria hepática inducida por fármacos puede considerarse predecible (alta incidencia) o impredecible (baja incidencia)66. La alteración en las pruebas hepáticas puede ser consecuencia de la toxicidad directa de la droga o de sus metabolitos, o puede ser una respuesta idiosincrásica en personas con una predisposición genética característica. El período de latencia entre el inicio del tratamiento y la aparición de la enfermedad en el hígado proporciona pistas claves sobre su etiología. Las reacciones hepatotóxicas prevenibles dependientes de la dosis, como es el caso de la intoxicación por paracetamol y su aparición temprana, son una evidencia consistente sobre del efecto directo del fármaco, sobre todo si no existe exposición previa66. Las reacciones hepatotóxicas no predecibles, no están relacionadas con la dosis, pero sí, con las características del huésped67. Sin embargo, la gran mayoría de las reacciones a fármacos son impredecibles. Estas se producen cuando el fármaco se transforma en un metabolito tóxico directo (metabolismo mediados por el huésped) o a través de la formación de un metabolito que sea capaz de montar una respuesta inmunológica (reacción de hipersensibilidad).
Sin embargo, independiente de las vías descritas, pueden coexistir más de un mismo mecanismo de toxicidad en un mismo individuo. Las diferencias en el metabolismo de los antiretrovirales puede llevar a un exceso de metabolitos potencialmente dañinos cuando los polimorfismos genéticos afectan las enzimas que los metabolizan68. El tiempo de aparición de la lesión puede ser larga (de 2 a 12 meses), lo que plantea problemas para la monitorización del paciente69. Estas vías metabólicas complejas pueden explicar lesiones hepáticas observadas con el uso de INNTR e IP70,71.
Algunos medicamentos pueden potenciar la activación de los receptores de muerte de células T y/o vías de estrés intracelular, dando lugar a un aumento del estrés oxidativo72. En respuesta, los hepatocitos promover mecanismos de citoprotección, tales como la formación de proteínas de choque térmico, que protegen el hígado contra metabolitos tóxicos68. Esta respuesta citoprotector puede explicar la normalización espontánea de las enzimas hepáticas que pueden ocurrir a pesar del mantenimiento de la terapia ARV (u otros medicamentos, tales como isoniazida). Por otra parte, el aumento y disminución de las transaminasas en sangre, después de la iniciación de los medicamentos puede estar relacionado con un fenómeno de “adaptación”, por lo que las pruebas de función hepática se normalizan a pesar de la exposición al fármaco en curso73.
PERFIL METABÓLICO Y ARVSLos pacientes con infección VIH presentan un incremento del riesgo cardiovascular (RCV) no sólo relacionado con los factores de riesgo tradicionales, sino con otros factores, tales como: la inflamación crónica causada por el VIH, la inmunodeficiencia y la posible acción directa de ciertos antirretrovirales. La relevancia clínica de la dislipidemia en el manejo de los pacientes con infección VIH radica en el hecho de que se trata de un factor de riesgo cardiovascular de primer orden, en parte modificable74.
Existen determinados ARV que se asocian con un mejor impacto metabólico sobre otros, encontrando variaciones incluso dentro de una misma familia. Dentro de la familia de los INNTR, NVP se asocia con un mejor perfil lipídico que EFV, en especial por su aumento en los niveles de colesterol HDL (HDLc). Este efecto esta mediado por la capacidad de NVP de incrementar la producción de Apo-A1. ETR y RPV también poseen un mejor perfil lipídico que EFV, sobre todo en lo referido a los Trigliceridos (TG) y colesterol total (CT), con similares efecto sobre HDLc. Entre los IP, LPV/r se asocia con mayores incrementos de TG y CT que el resto de los fármacos de su clase. La nueva familia de los INSTI se asocia a un perfil más neutro, siendo superior al de EFV en cuanto a TG y CT. MVC posee también un perfil lipídico neutral y favorable que el de EFV.
Dentro de la familia de INTR, los fármacos que se asocian con peor perfil lipídico son ZDV y d4T, con mayor probabilidad de hipertrigliceridemia y elevación de CT y LDLc que el resto de los fármacos de esta familia. 3TC presenta un efecto neutro sobre los lípidos. TDF se asocia con un menor incremento de CT, LDLc y TG que ABC75
Es por ello que previo al inicio de la TAR se debe valorar el riesgo cardiovascular de los pacientes, para elegir el tratamiento más adecuado desde el punto de vista metabólico. La justificación de modificar la TAR antes del uso de hipolipemiantes debe ser analizado y valorado individualmente, siempre que no exista riesgo de falla virológica y sin olvidar considerar que el paciente se expone a los efectos adversos del nuevo fármaco.
El autor declara no tener conflictos de interés, en relación a este artículo.