



We report the case of a 43-year-old male smoker who presented to the emergency department with skin lesions resembling pyoderma on the lower extremities, punctate and necrotic digital ulcers, fever, malaise, polyarthralgia and elevated inflammatory markers. He later developed bilateral, asymptomatic cavitary pulmonary nodules, diffuse alveolar hemorrhage, pansinusitis, and positive PR3-ANCA, establishing the diagnosis of GPA. An excellent initial response was observed with pulse methylprednisolone, oral prednisolone, intravenous pulse cyclophosphamide, and hyperbaric oxygen therapy (HOT).
Three months later, however, he was diagnosed with a NET of the small intestine. It was polypoid and multifocal, with high catecholamine production but low Ki-67, leading to a reduction in immunosuppressive therapy. Two new flares occurred, with skin, renal, ocular, intestinal, and joint involvement, along with elevated c-ANCA levels, prompting a reinduction strategy with rituximab, plasmapheresis, corticosteroids, and HOT, resulting in significant improvement.
In addition to the complex and challenging clinical management of this case, this unique combination – GPA and NET – is highlighted.
Informamos el caso de un hombre de 43 años, fumador, que se presentó en el departamento de emergencias con lesiones cutáneas en las extremidades inferiores, similares a pioderma, úlceras digitales puntiformes y necróticas, fiebre, malestar, poliartralgia y marcadores inflamatorios elevados. Posteriormente, desarrolló nódulos pulmonares cavitarios bilaterales asintomáticos, hemorragia alveolar difusa, pansinusitis y ANCA PR3 positivo, estableciéndose el diagnóstico de GPA. Se observó una excelente respuesta inicial con pulsos de metilprednisolona, prednisolona oral, pulsos intravenosos de ciclofosfamida y terapia de oxígeno hiperbárico (TOH).
Sin embargo, tres meses después, se diagnosticó un TNE del intestino delgado, de tipo polipoide y multifocal, con alta producción de catecolaminas pero bajo Ki-67, lo que llevó a una reducción de la terapia inmunosupresora. Ocurrieron dos nuevos brotes con afectación cutánea, renal, ocular, intestinal y articular, junto con niveles elevados de c-ANCA, lo que requirió una estrategia de reinducción con rituximab, plasmaféresis, corticosteroides y TOH, logrando una mejoría significativa.
Además de la complejidad y el desafío en el manejo clínico de este caso, se destaca esta combinación única de GPA y NET.
Granulomatosis with polyangiitis (GPA), is an autoimmune disease of unknown etiology, characterized by small-vessel vasculitis with granulomatous inflammatory lesions and necrosis. GPA is part of the spectrum of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitides, which also includes eosinophilic granulomatosis with polyangiitis and microscopic polyangiitis1. The cytoplasmic pattern (c-ANCA), primarily associated with antibodies against proteinase-3 (PR3), has high specificity, being present in about 90% of patients and is more commonly associated with generalized disease. Upper and lower respiratory tract and renal involvement are the most common features1,2.
Of the three main ANCA-associated vasculitides, GPA is the most common, although rare, with an estimated prevalence in Europe and America of 120-140 per million inhabitants, with a higher incidence in Northern Europe. It affects males and females equally, occurring predominantly between the ages of 45 and 60, although a small proportion (3-7%) may affect children and adolescents3.
In 2022, new classification criteria for GPA were published by the American College of Rheumatology (ACR)/European Alliance of Associations for Rheumatology (EULAR). These criteria have high sensitivity and specificity when applied to patients with small-vessel vasculitis. They include clinical, laboratory, imaging, and biopsy criteria, requiring at least 5 points for classification as GPA3.
The main treatment for GPA is a combination of corticosteroids and cytotoxic agents. Treatment must be tailored to appropriately manage GPA manifestations while minimizing long-term toxicity. Untreated GPA has a very poor prognosis, with an average life expectancy of five months and a one-year survival rate of less than 20%1,4.
Disease outcomes improved significantly with the introduction of cyclophosphamide, administered in combination with corticosteroids. Approximately 90% of GPA patients respond to cyclophosphamide, with about 75% achieving complete remission. However, 30%-50% experience at least one relapse. The FDA has also approved rituximab, an anti-CD20 monoclonal antibody, as an alternative to cyclophosphamide. Currently, both can be used as first-line therapy in severe disease, with rituximab being preferred for non-life-threatening disease1,4.
Neuroendocrine tumors (NETs) are a rare and heterogeneous group of neoplasms originating from neuroendocrine cells, most commonly occurring in the digestive system, followed by the lungs. Among gastrointestinal NETs, primary lesions are most frequently found in the gastric mucosa, small and large intestine, rectum, and pancreas5,6.
NETs can be subdivided into functional and non-functional. Functional NETs overproduce endogenous hormones or vasoactive substances, potentially resulting in significant clinical repercussions. These tumors can arise sporadically or in the context of a hereditary tumor syndrome6. Their prevalence increases from the fifth decade of life, with a slight predominance in males. In Europe, the incidence of gastrointestinal NETs is about 1.33-2.33/100 000 inhabitants5.
Histological diagnosis is mandatory, as all tumors share a common phenotype with immunoreactivity for so-called ‘pan-neuroendocrine’ markers, including chromogranin A and synaptophysin. Immunohistochemistry for Ki-67, a proliferation index, is essential for classification according to the World Health Organization (WHO) (G1 Ki-67 =3%; G2 3-20%; G3 >20%). The tumor's location, Ki-67, and local and distant involvement are crucial for prognosis. Surgical treatment is recommended if the tumor is larger than 2cm or in presence of local invasion5.
Case reportWe present the case of a 43-year-old male, with no known personal or family medical history, who was referred to the emergency department due to the appearance of punctate skin lesions that within a two-week evolution rapidly progressed to larger necrotic ulcers on the right leg. In the 48hours prior to admission, the patient developed fever, polyarthralgia and punctuated necrotic lesions on the nails and palms of his hands. The patient denied Raynaud's phenomenon, weight loss, diarrhea, headaches, nasal changes, jaw claudication, dyspnea, cough, hemoptysis, or peripheral edema.
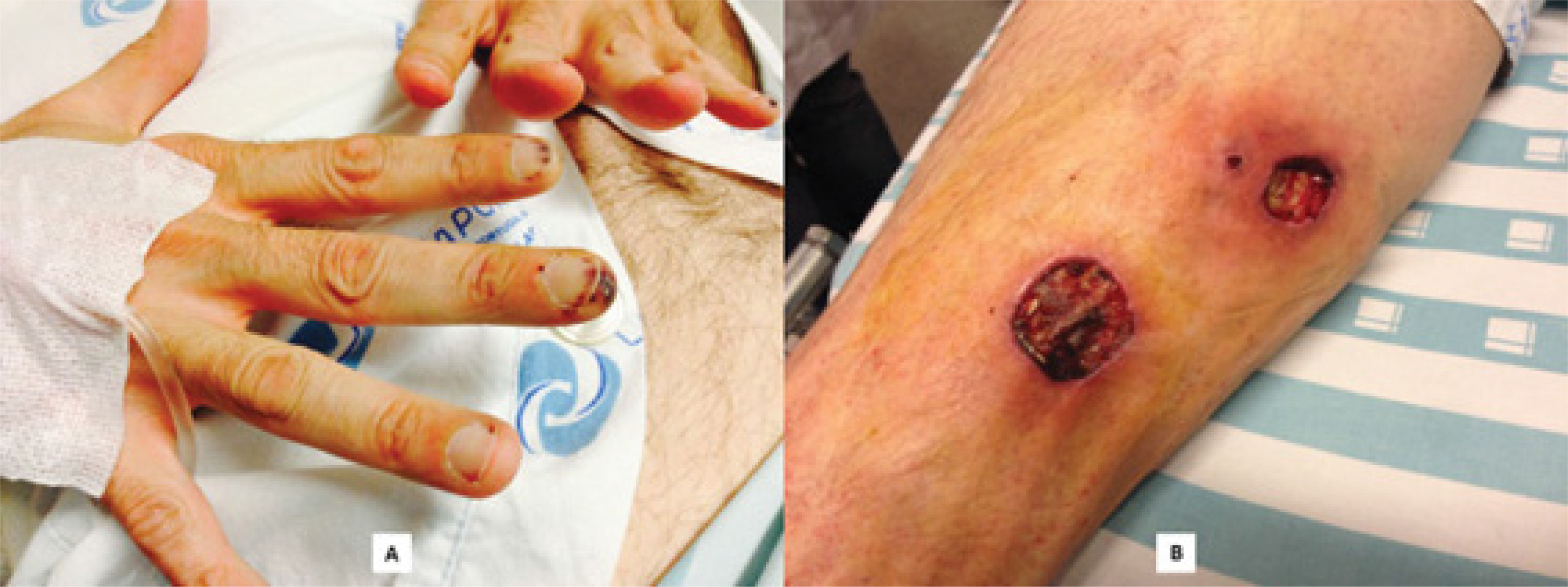
On physical examination, the patient presented with fever (38°C), no neurological deficits and a normal cardiorespiratory examination. He had two necrotic lesions resembling pyoderma on the right leg and bilateral punctate violaceous lesions on palms and fingers (figure 1).
 and large necrotic ulcers in the right leg (B).")
Laboratory exams revealed leukocytosis with neutrophilia, without eosinophilia, elevated C-reactive protein (CRP) (28.93mg/dl), elevated erythrocyte sedimentation rate (ESR) (103mm/h), normal kidney function, and microhematuria without proteinuria. A chest X-ray showed a nodular infiltrate in the left hemithorax.
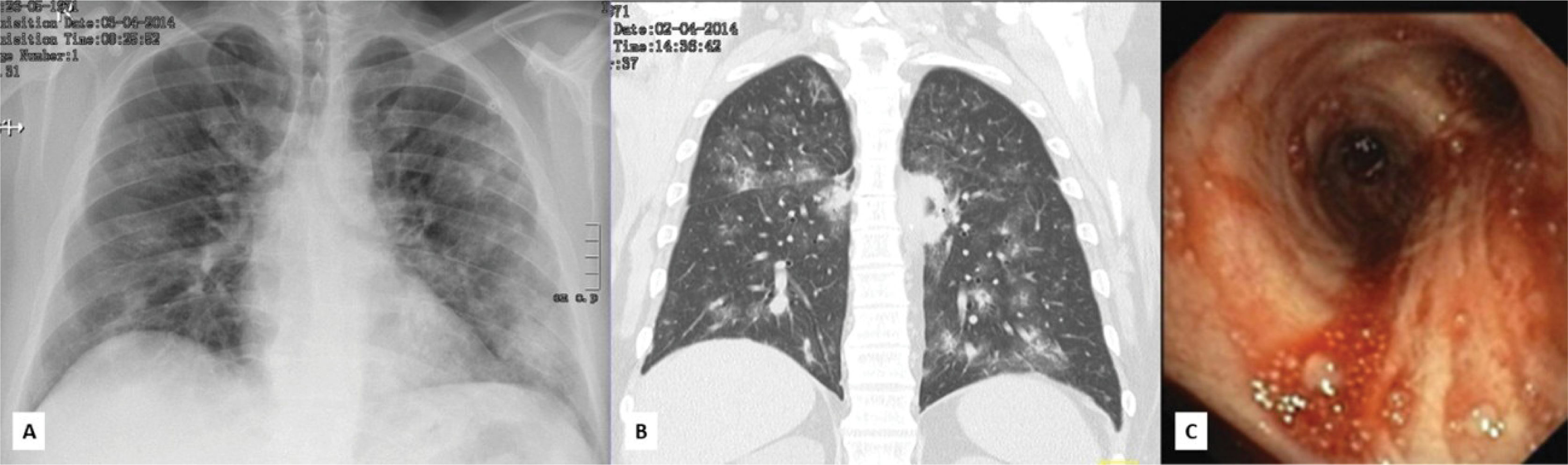
The differential diagnosis included endocarditis, sepsis with a respiratory focus, pyoderma gangrenosum, and vasculitis. Antibiotic therapy was initiated, and further laboratory assessment was requested. Seventy-two hours after admission, a drop of 2g in hemoglobin was observed and a thoracoabdominal computed tomography (CT) was performed, revealing bilateral cavitating pulmonary nodules, as well as reticular densification in the lower lobes, suggestive of alveolar hemorrhage, later confirmed by bronchoscopy (figure 2) and bronchoalveolar lavage. Direct and culture tests for mycobacteria and fungi were negative. The abdominal CT was normal. Transthoracic and transesophageal echocardiograms ruled out vegetations. Further laboratory data revealed positive PR3-ANCA (148 U/ml; Normal <7 U/ml). A perinasal CT scan showed pansinusitis, and skin biopsy with indirect immunofluorescence was non-specific.
 and CT (B); pulmonary hemorrhage in bronchoscopy (C).")
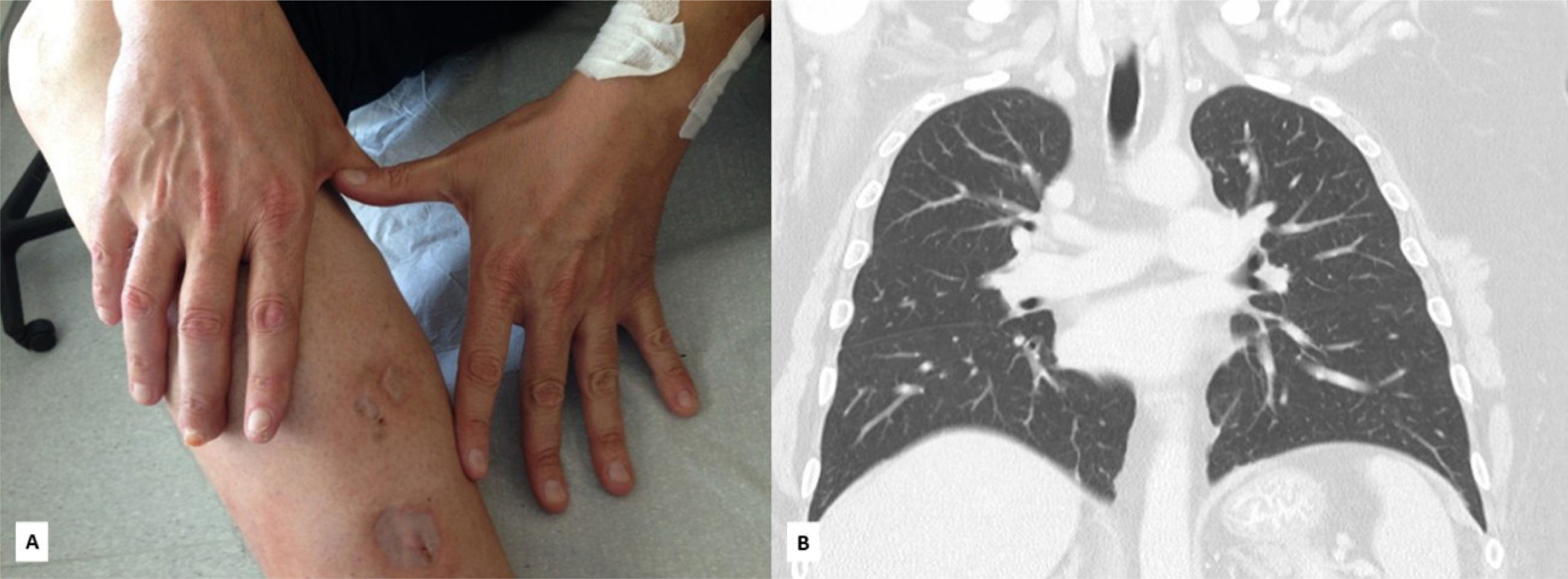
The diagnosis of generalized GPA was established based on skin and respiratory involvement and highly positive PR3-ANCA. Renal involvement at the time was underestimated by nephrology due to mild microhematuria and the absence of significant proteinuria or renal dysfunction. Immunosuppressive therapy with methylprednisolone (1g IV/day for 3 days), followed by prednisolone (1mg/kg/day) and IV pulse cyclophosphamide (750mg/m2/month) was initiated. Complementary therapy with daily local dressings, vasodilators, and hyperbaric oxygen therapy (HOT) sessions resulted in excellent skin improvement. The ANCA titer normalized, and the pulmonary nodules and alveolar hemorrhage regressed (figure 3).
 and pulmonary cavitations (B).")
Three months later, while still on prednisolone 20mg/day and monthly cyclophosphamide cycles, the patient returned to the emergency department with dyspnea, high fever, and anorexia, evolving for one week. There were no abdominal complaints, but the patient reported a low-impact car accident the previous week.
On physical examination, the patient had fever and tachypnea, but cardiorespiratory auscultation and the abdominal exam were normal, with significant skin improvement. Laboratory tests showed inflammatory anemia (Hb 10.5g/dl), CRP of 11.52mg/dl, LDH of 633 IU/l, and hypoxemia with hypocapnia (pCO2 27mmHg, pO2 53mmHg, and lactate 6mg/dl). A thoraco-abdominal CT angiography ruled out pulmonary embolism and revealed improvement in pulmonary cavitations but detected multinodular thickening involving the lesser curvature and the gastric antrum, body and tail of the pancreas, and proximal jejunum. Serum amylase and lipase, as well as IgG4, were normal.
The upper endoscopy revealed two polyps in the gastric bulb and body, which were excised; endoscopic ultrasound detected nodular thickening between the gastric wall, pancreas, and jejunal loops (figure 4). Polyp histology showed an adenoid and trabecular pattern (positive for chromogranin and synaptophysin, low Ki-67-3%), suggestive of a NET. Histology of transgastric aspiration was consistent with neuroendocrine origin. The patient had no family history of NET and had a negative genetic test (tested outside our hospital).
Further investigation revealed elevated levels of chromogranin A, normetanephrine, and gastrin, without clinical evidence of a functional syndrome. Scintigraphy with 123-metaiodobenzylguanidine (MIBG) excluded pheochromocytoma or paraganglioma, and a positron emission tomography (PET) scan with gallium DOTA NOC showed a slight uptake in the duodenum. In addition, balloon-assisted enteroscopy revealed 3 polyps (also NETs) in the duodenal bulb and serpiginous ulcers, along with volcano-like aphthous lesions in the small intestine. Biopsy showed thromboangiitis and lymphocytic infiltration, suggestive of vasculitis.
The diagnosis of multifocal, low-grade, polypoid NET was made in a patient with ANCA-positive small vessel vasculitis, also with intestinal involvement, and undergoing induction therapy. Due to the neoplastic process, early reduction (in the 5th cycle) of cyclophosphamide to methotrexate and a gradual tapering of prednisolone were decided, along with active imaging surveillance, as surgical intervention was not an option due to the tumor's multifocal nature.
However, 4 months later, a new exacerbation occurred, with the reappearance of skin lesions on the limbs, resembling pyoderma, polyarthritis, anterior uveitis in the left eye, renal dysfunction, hematuria-proteinuria (900mg/24h), and elevated PR3-ANCA titers (107 U/l). Due to the overall severity, the renal biopsy was postponed. In this context, rituximab (375mg/m2/week for 4 weeks), IV pulse methylprednisolone, plasmapheresis, and adjuvant HOT was initiated, resulting in a notable, though slow, improvement of the vasculitis. Interestingly, the NETs regressed over time.
After 9 months of disease inactivity, under maintenance with azathioprine (125mg/day) and prednisolone 2.5mg, there was another exacerbation (pulmonary, cutaneous, articular, and renal), with worsening nephrotic-range proteinuria and renal function. A renal biopsy was performed, consistent with pauci-immune crescentic glomerulonephritis, with four sclerosed glomeruli and 75% interstitial fibrosis and tubular atrophy. A multidisciplinary decision was made to administer rituximab cycles over 9 months.
Discussion and conclusionThis patient presented an ANCA-associated vasculitis with multiorgan involvement. Approximately half of patients with ANCA-associated vasculitis exhibit cutaneous manifestations, primarily leukocytoclastic vasculitis, which causes purpura in the lower limbs with focal ulceration and necrosis7.
Despite a good response to induction therapy with both cyclophosphamide and rituximab, maintenance therapy was more challenging. The diagnosis and treatment of ANCA-associated vasculitis are associated with an increased risk of de novo malignancies, due to compromised immune surveillance, the direct oncogenicity of immunosuppressives, and possibly the malignant degeneration of tissues subjected to chronic immune stimulation8,9.
Although immunosuppressive therapy was crucial in this case, we highlight the central role of HOT in the excellent outcome, which was initiated once the pulmonary lesions improved. HOT enhances tissue oxygenation, leading to fibroblast proliferation, collagen synthesis and growth factor production. It also enhances neutrophil-mediated bacterial destruction, promoting ulcer healing10.
Plasma exchange was used for rapidly progressive glomerulonephritis. Following the renal biopsy, a poor renal prognosis was confirmed, and the patient is currently undergoing renal replacement therapy while awaiting a transplant.
The diagnosis of malignancy presented an additional challenge. Despite the risks, given the severe exacerbations, it was necessary to initiate new induction therapy, in accordance with updated guidelines recommending rituximab. According to the literature, the risk of malignancy is higher in patients with ANCA-associated vasculitis compared to the general population, with a possibly greater risk in GPA than in other ANCA-associated vasculitides8,9. Cyclophosphamide has been identified as a major contributor to the development of malignancies due to its direct carcinogenic properties8. Conversely, cancer has also been suggested as a potential trigger of ANCA-associated vasculitis. It can be speculated in this case whether ANCA-associated vasculitis was the cause or consequence of the neuroendocrine tumors (NETs), possibly as a paraneoplastic syndrome. However, the NET was of low grade, and the vasculitis exhibited typical features.
No cases of association between ANCA-associated small vessel vasculitis and gastrointestinal NETs have been found in the literature, confirming the rarity and complex challenge of managing this seemingly antagonistic combination of cancer and vasculitis.
Ethics StatementThe patient has provided their informed consent for the writing and publication of this article.
Conflict of Interest StatementThere are no conflicts of interest.
FundingThis research has not received specific aid from public sector agencies, the commercial sector or non-profit entities.