





La hipertensión arterial pulmonar (HAP) es una condición rara con presión arterial pulmonar elevada (>20mmHg) y resistencia vascular aumentada. Afecta entre 5 y 15 adultos por millón, y provoca disnea, dolor torácico, y riesgo de insuficiencia cardíaca derecha y muerte. A continuación, se describe un caso de presentación grave de hipertensión arterial pulmonar cuyo diagnóstico se establece luego de un paro cardíaco recuperado.
Caso clínicoHombre de 48 años que consulta por edemas, presíncope y disnea progresiva de 6 meses de evolución. Sufre paro cardiorrespiratorio por actividad eléctrica sin pulso y luego asistolia, requiriendo reanimación, noradrenalina y ventilación mecánica. El ecocardiograma muestra hipertensión pulmonar severa (presión sistólica arteria pulmonar 59mmHg) con función ventricular izquierda conservada y dilatación severa del ventrículo derecho. La angiotomografía descarta tromboembolismo pulmonar. Se instaura tratamiento con dobutamina y balance negativo, con mejoría hemodinámica. El cateterismo cardíaco derecho confirma hipertensión pulmonar precapilar. Se inician sildenafil, óxido nítrico y posteriormente, ambrisentán. Los estudios reumatológicos revelan artritis reumatoide con sobreposición Sjögren como probable etiología de la hipertensión arterial pulmonar. El paciente mejora con tratamiento dirigido a su hipertensión pulmonar y prednisona, lográndose suspender ventilación mecánica y darse de alta, manteniéndose estable al mes de seguimiento.
DiscusiónLa HAP asociada a enfermedades del tejido conectivo, como el síndrome de Sjögren y la artritis reumatoide, es poco prevalente. Los tratamientos incluyen antagonistas de los receptores de endotelina, inhibidores de fosfodiesterasa-5, prostanoides y combinaciones de fármacos. El tratamiento inmunológico adecuado de las enfermedades reumatológicas subyacentes puede mejorarla. En el presente caso clínico, el manejo integral, incluyendo vasodilatadores pulmonares y control de la precarga, fue clave para la recuperación del paciente.
Pulmonary arterial hypertension (PAH) is a rare condition characterized by elevated pulmonary artery pressure (>20mmHg) and increased vascular resistance. It affects 5 to 15 adults per million, causing dyspnea, chest pain, risk of right heart failure and death. Below, we describe a case of severe pulmonary arterial hypertension whose diagnosis was established after a recovered cardiac arrest.
Clinical CaseA 48-year-old man presented with edema, presyncope, and progressive dyspnea over 6 months. He suffered a cardiorespiratory arrest due to pulseless electrical activity and subsequent asystole, requiring resuscitation, norepinephrine, and mechanical ventilation. Echocardiography revealed severe pulmonary hypertension (pulmonary artery systolic pressure of 59mmHg), preserved left ventricular function, and severe right ventricular dilation. Computed tomography angiography ruled out pulmonary embolism. Treatment with dobutamine and negative fluid balance improved his hemodynamics. Right heart catheterization confirmed precapillary pulmonary hypertension. Sildenafil, nitric oxide, and later, ambrisentan were initiated. Rheumatologic studies identified rheumatoid arthritis with overlapping Sjögren syndrome as the likely etiology of his pulmonary hypertension. The patient improved with targeted pulmonary hypertension therapy and prednisone, was successfully weaned off mechanical ventilation and was discharged, remaining stable at the one-month follow-up.
DiscussionPAH associated with connective tissue diseases, such as Sjögren syndrome and rheumatoid arthritis is rare. Treatments include endothelin receptor antagonists, phosphodiesterase-5 inhibitors, prostanoids, and drug combinations. Appropriate immunological treatment for underlying rheumatologic diseases, like rheumatoid arthritis, may improve PAH. In this case, an integral approach, including pulmonary vasodilators and preload control, was key to the patient's recovery.
La hipertensión arterial pulmonar (HAP), es una condición clínica poco frecuente, caracterizada por un aumento anormal de la presión en la arteria pulmonar mayor a 20mmHg, resistencia vascular pulmonar (RVP) =2 unidades Wood y presión capilar pulmonar de enclavamiento (PCP) =15mmHg1. En la población general, se estima que hay entre 5 y 15 casos de hipertensión pulmonar del grupo I por cada millón de adultos. Esta condición se manifiesta principalmente con disnea, dolor torácico, síncope, pudiendo progresar a insuficiencia cardíaca derecha y muerte si no se trata adecuadamente.
A continuación, se describe un caso de presentación grave de hipertensión arterial pulmonar cuyo diagnóstico se establece luego de un paro cardíaco recuperado.
Caso clínicoPaciente de sexo masculino de 48 años que consultó en otra institución por un cuadro de seis meses de evolución caracterizado por edemas, episodios presincopales y disnea progresiva hasta capacidad funcional IV. Evolucionó con paro cardiorrespiratorio (PCR) por actividad eléctrica sin pulso y luego asistolia. Se realizó reanimación cardiopulmonar avanzada con posterior retorno a la circulación espontánea en ritmo sinusal, con requerimiento de noradrenalina y ventilación mecánica invasiva. Es derivado a nuestra clínica para continuar con estudio y tratamiento.
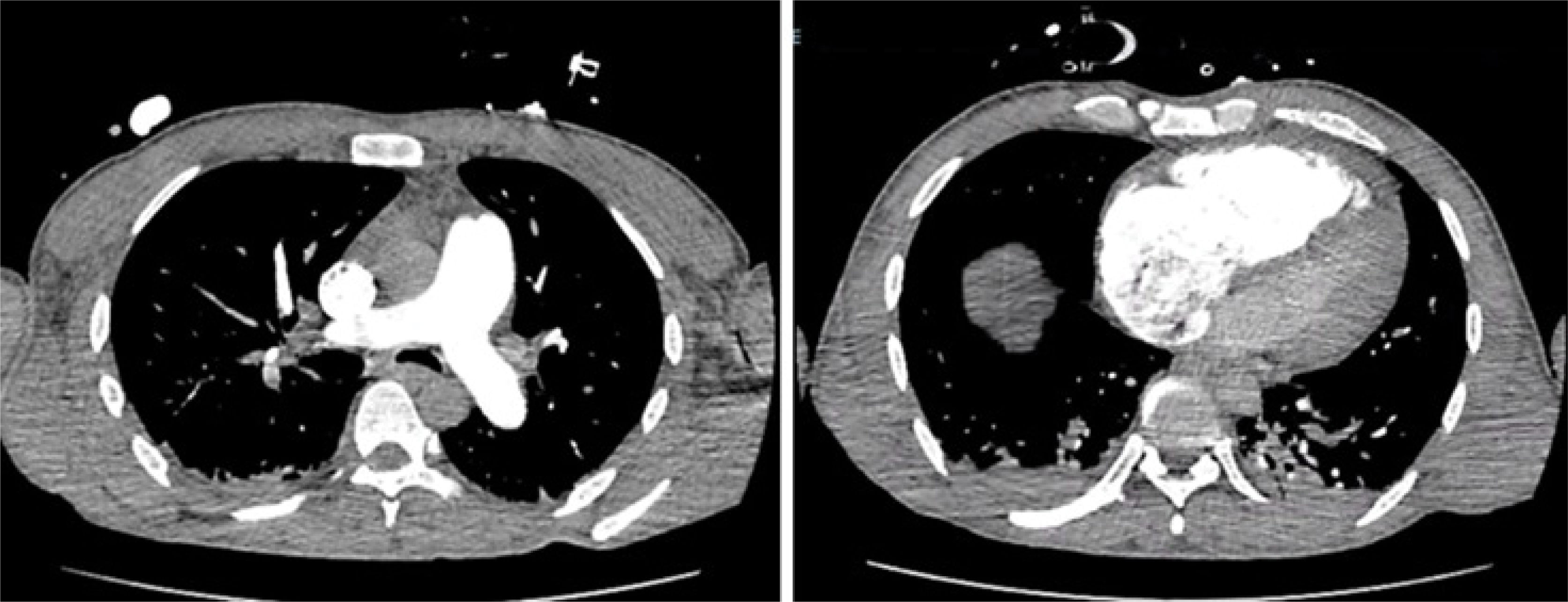
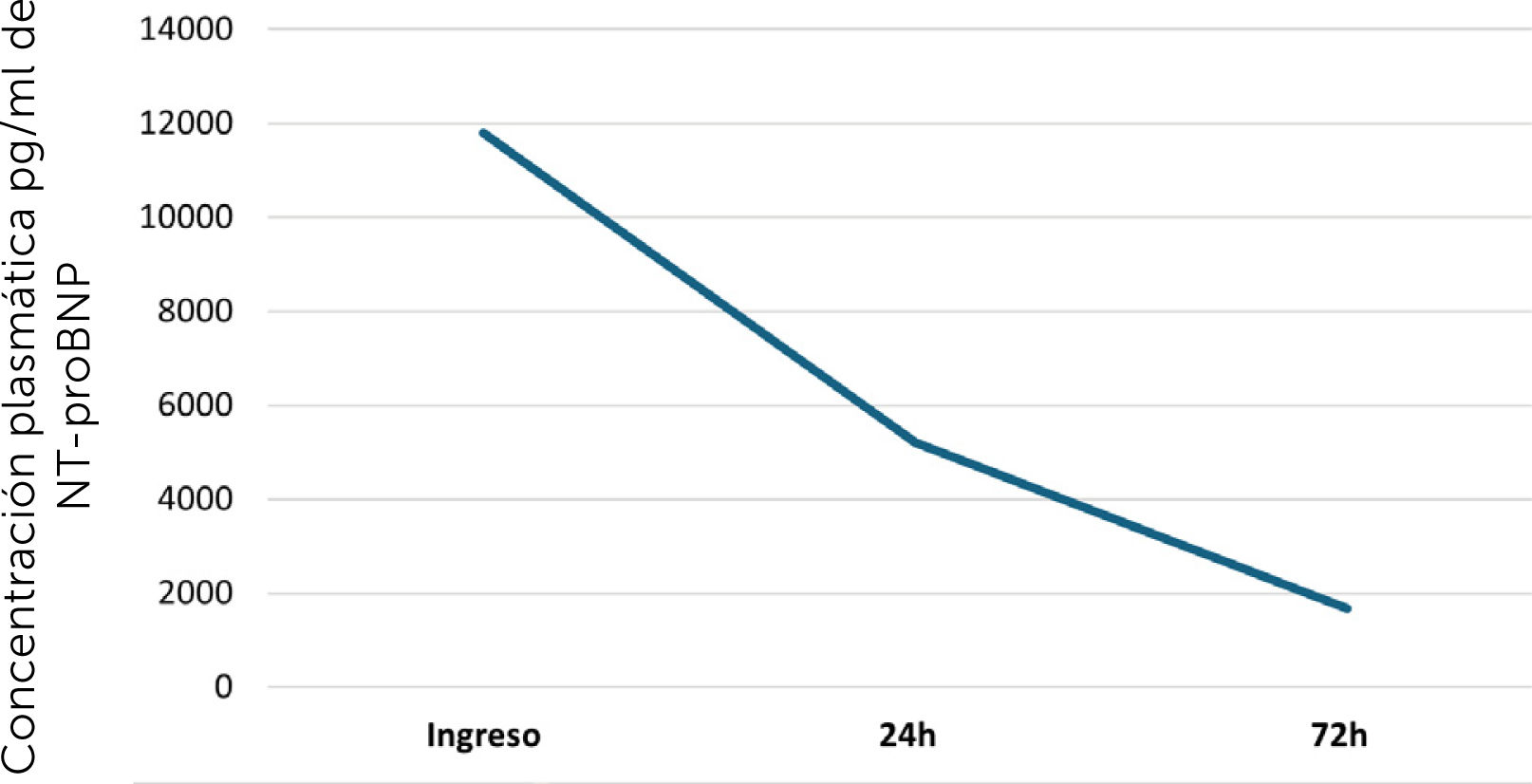
Ingresó a nuestra institución sedado, en ventilación mecánica, con noradrenalina hasta 0,4μg/kg/min y vasopresina hasta 0,03μg/min, destacando al examen físico edema generalizado de extremidades y crépitos gruesos en ambos campos pulmonares. El primer día fue evaluado mediante ecocardiograma transtorácico destacando una función ventricular izquierda conservada, signos de hipertensión pulmonar severa con una estimación de presión de arteria pulmonar sistólica (PAPS) de 59mmHg, dilatación severa de cavidades derechas con disfunción ventricular derecha y dilatación de vena cava inferior (30mm) con ausencia de colapso inspiratorio. Se realizó también angiotomografía pulmonar con contraste, sin evidencia de tromboembolismo pulmonar ni alteraciones del parénquima pulmonar y con relación ventrículo derecho/ventrículo izquierdo aumentada, mayor a 1 (figura 1). Se interpretó el cuadro como shock cardiogénico por falla ventricular derecha secundaria a hipertensión pulmonar, por lo que se inició dobutamina hasta 6μg/kg/min y furosemida endovenosa (EV) para lograr balance hídrico negativo. Paralelamente, se inició óxido nítrico inhalado 20ppm y sildenafil 50mg cada 8h. El paciente evolucionó favorablemente, logrando suspensión de vasopresores. A las 48h de su ingreso, se realizó cateterismo derecho (bajo el tratamiento vasodilatador pulmonar mencionado) mediante catéter de Swan-Ganz que evidencia hipertensión pulmonar con resistencia vascular pulmonar elevada y presión capilar pulmonar normal. Se logra descenso progresivo de dobutamina, realizándose nueva medición de presiones pulmonares (tabla 1). Paralelamente, se solicitó estudio completo para hipertensión pulmonar precapilar incluido perfil inmunológico que evidencia patrón compatible con artritis reumatoide (AR) y sobreposición de Sjögren (tabla 2). Al interrogar a los familiares, estos refieren que el paciente desde hace aproximadamente un año presenta dolores articulares en ambas muñecas. Bajo ese contexto, se inició prednisona 0,5mg/kg/día. Paralelamente, se inició ambrisentán 5mg/día vía oral que posteriormente se titula a 10mg/día. El paciente evolucionó favorablemente, logrando desconexión de ventilación mecánica invasiva y peso seco tras balance hídrico negativo forzado con furosemida, acompañado de descenso progresivo de valores de NT-proBNP (figura 2). Se realiza ecoscopía al día 23 de su ingreso, tras 20 días de sildenafil, 18 días de ambrisentán y en peso seco sin intotrópicos, que revela función ventricular derecha conservada TAPSE (por sus siglas en inglés de tricuspid annular plane systolic excursion) de 26mm de diámetro, ventrículo derecho de 42mm, fracción de eyección ventricular izquierda 65% con una PAPS de 30mmHg, lográndose finalmente el alta hospitalaria.En el seguimiento por consultorios externos al mes del alta, el paciente no ha presentado nuevas descompensaciones y continúa con controles con reumatología y cardiología.
.")
Mediciones de cateterismo derecho mediante catéter de Swan-Ganz (48 h y 7 días luego del ingreso)
| Parámetro | 48 horas | 7 días |
|---|---|---|
| Frecuencia cardíaca (lpm) | 71 | 64 |
| Presión arterial media (mmHg) | 62 | 73 |
| Presión sistólica/diastólica/media de arteria pulmonar (mmHg) | 40/21/30 | 35/18/24 |
| Presión capilar pulmonar (mmHg) | 10 | 8 |
| Volumen minuto (l/min) | 6,4 | 6 |
| Índice cardíaco (l/min/m2) | 3,3 | 3,1 |
| Resistencia vascular pulmonar (UW) | 3 | 2,66 |
Perfil reumatológico a las 48 h de ingreso
| Factor reumatoideo | >900UI/ml |
| Anti-CCP | >200UI/ml |
| ANA | > 1/640 |
| Anti-Ro | > 60UI/ml |
| C3 y C4 | Disminuidos |
Abreviatura: UI/ml: unidades internacionales por mililitro; anti-CCP: anticuerpos antipéptido citrulinado; ANA: anticuerpos antinucleares; Anti-Ro o SSA: (por sus siglas en inglés de anti-Sjögren's syndrome related antigen A).

La HAP asociada a enfermedades del tejido conectivo forma parte de la hipertensión pulmonar del grupo I. Aquella asociada con un síndrome de Sjögren primario y artritis reumatoide es una entidad rara pero grave2,3. La patogenia de la enfermedad asociada a estas dos condiciones clínicas es poco clara, sin embargo se ha identificado que las mutaciones en el gen del receptor de la proteína morfogenética ósea tipo 2 (BMPR2) están presentes en muchos de estos casos, lo que sugiere una predisposición genética4,5. Además, se han propuesto varias teorías que implican una proliferación y remodelación vascular en las pequeñas arterias pulmonares, con hipertrofia de la media, fibrosis de la íntima, microtrombosis y lesiones plexiformes; lo que llevaría a un aumento progresivo de la resistencia vascular pulmonar y, en última instancia, a la insuficiencia ventricular derecha y la muerte1,6,7.
El diagnóstico de HAP del grupo I requiere excluir hipertensión pulmonar secundaria a patología cardíaca izquierda, trastornos pulmonares y obstrucciones de la arteria pulmonar. Para ello, es fundamental realizar el cribado completo ante la presencia de hipertensión arterial pulmonar: ecocardiograma con test de burbujas, angiotomografía de tórax de corte fino, ecodoppler portal, espirometría, la prueba de difusión de monóxido de carbono (DLCO), serologías para VIH, VHC y VHB, analítica completa, perfil renal, perfil inmunológico: C3, C4, ANA, anti-DNA, perfil ENA, factor reumatoideo, péptido citrulinado, anti-cardiolipinas, anti-β2-glicoproteína y anticoagulante lúpico. La ecocardiografía es una herramienta no invasiva fundamental para la sospecha inicial, pero la confirmación del diagnóstico y la evaluación hemodinámica requieren cateterismo cardíaco derecho1. Durante el cateterismo, se debe realizar una prueba de vasorreactividad en los siguientes pacientes con HAP del grupo I; HAP idiopática, HAP hereditaria y HAP asociada a enfermedad del tejido conectivo. La prueba se realiza idealmente con óxido nítrico o prostanoide inhalado. Los criterios de respuesta positiva son: disminución de presión media de la arteria pulmonar (PAPm) mayor o igual a 10mmHg y PAPm final menos de 40mmHg sin disminución del gasto cardíaco. Aproximadamente el 10% de los pacientes con HAP idiopática responden favorablemente a esta prueba. Estos pacientes son candidatos a tratamiento con bloqueantes cálcicos a largo plazo. Una prueba negativa indica que los pacientes no se beneficiarán del tratamiento con bloqueantes cálcicos, y deben considerarse otras terapias específicas para HAP (antagonistas de receptores de endotelina, inhibidores de la fosfodiesterasa-5, prostanoides).
El tratamiento de la HAP ha avanzado significativamente en las últimas décadas. Las terapias actuales incluyen:
- 1.
Antagonistas de los receptores de endotelina (ARE): medicamentos como bosentán, ambrisentán y macitentán han demostrado eficacia en la reducción de la resistencia vascular pulmonar y la mejora de la capacidad funcional en pacientes con HAP8.
- 2.
Inhibidores de la fosfodiesterasa tipo 5 (PDE5i): fármacos como sildenafil y tadalafilo mejoran la vasodilatación pulmonar y han mostrado beneficios en la capacidad de ejercicio y la hemodinamia pulmonar9.
- 3.
Prostanoides: incluyen epoprostenol, treprostinil e iloprost, que son potentes vasodilatadores pulmonares y tienen efectos antiproliferativos1. Estos agentes pueden administrarse por vía intravenosa, subcutánea o inhalada, dependiendo de la situación clínica del paciente.
- 4.
Estimuladores de la guanilato ciclasa soluble: riociguat es un agente que ha mostrado eficacia en la mejoría de la capacidad de ejercicio y la hemodinamia en pacientes con HAP6.
- 5.
Terapias combinadas: La combinación de diferentes clases de medicamentos, como un ARE con un PDE5i, ha demostrado ser más efectiva que la monoterapia en mejorar los resultados clínicos y la supervivencia1,8.
Existen casos en que el tratamiento de una enfermedad reumatológica subyacente puede mejorar o estabilizar la HAP. La mejoría de la HAP secundaria a enfermedades reumatológicas es posible cuando el tratamiento inmunológico es oportuno y adecuado, siempre y cuando la HAP esté relacionada con inflamación o trombosis activa10.
En el paciente presentado, parte de la disfunción ventricular derecha podría haberse debido a la reanimación con cristaloides recibida luego del paro cardíaco en otra institución en un paciente con HAP previa, lo que explicaría la normalización de la función del ventrículo derecho una vez sometido a balance negativo y habiéndose normalizado la poscarga con vasodilatadores pulmonares específicos y el tratamiento de su enfermedad reumatológica de base.
ConclusiónEn el caso de la AR, la mejoría de la HAP puede observarse con el manejo adecuado de la AR mediante terapias biológicas o inmunosupresores. En el caso particular de nuestro paciente, creemos que el abordaje integral fue clave en el desenlace favorable: inotrópicos, manejo de precarga con diuréticos, manejo de precarga con vasodilatadores pulmonares e inmunosupresión para artritis reumatoide activa.
Consideraciones éticasSe ha obtenido el consentimiento informado del paciente para la publicación de su información clínica.
Declaración de conflicto de interesesLos autores declaran que no tienen ningún conflicto de intereses relacionado con la publicación de este artículo.
Declaración de fuente de financiamientoEste estudio no ha recibido financiamiento externo.