




La hipertensión arterial (HTA) dependiente de mineralocorticoides representa actualmente una de las formas secundarias de hipertensión más prevalentes. Entre las causas más conocidas está el hiperaldosteronismo primario (HAP). A nivel renal, la aldosterona reabsorbe sodio y agua aumentando el volumen intravascular y la presión arterial. Actualmente la prevalencia de HAP, detectada por la razón aldosterona/actividad renina plasmática (ARR) que es considerado el mejor test de screening, es cercana al 10% en población hipertensa. Por otra parte, defectos congénitos o adquiridos en la enzima 11β-Hidroxiesteroide deshidrogenasa tipo 2 (11β-HSD2) resultan en una ineficiente inactivación de cortisol a cortisona favoreciendo la aparición de hipertensión por activación del receptor mineralocorticoideo. La actividad de esta enzima se evalúa midiendo la razón cortisol/cortisona en suero o en orina de 24 horas. Recientemente, hemos observado déficit parciales de la actividad de la enzima 11β-HSD-2 en alrededor del 15% de los pacientes hipertensos esenciales los que podrían ser tratados con bloqueadores específicos del receptor mineralocorticoide y/o con corticoides de acción prolongada sin actividad mineralocorticoide como dexametasona o betametasona.
Mineralocorticoid-dependent hypertension represents one of the most prevalent causes of secondary hypertension. Primary aldosteronism (PA) is a known cause of hypertension (HT). In the kidney, aldosterone increases reabsorption of sodium and water increasing intravascular volume and blood pressure. To date, the PA prevalence is about 10% in hypertensive population detected by the aldosterone / plasma renin activity ratio (ARR) which is considered the best screening test. Moreover, congenital or acquired defects in the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) results in an impaired inactivation of cortisol to cortisone leading to the development of hypertension by mineralocorticoid receptor activation. The activity of this enzyme is assayed by measuring the cortisol/cortisone ratio in serum or in urine of 24 hrs. Recently, we have observed partial deficiency of the 11β-HSD-2 enzyme activity in about 15% of the essential hypertensive patients who could be treated with mineralocorticoid receptor-specific blockers and/or long-acting corticosteroid without mineralocorticoid activity as dexamethasone or betamethasone.
Financiamiento: Proyectos FONDEF IDEA CA12I10150, FONDECYT 1130427, SOCHED 2012-04 & IMII P09/016-F. CAC es becado doctoral de la Comisión Nacional y Científica de Chile (CONICYT).
La hipertensión dependiente de mineralocorticoides representa actualmente una de las formas secundarias de hipertensión más prevalentes. La causa más común es el hiperaldosteronismo primario (HAP) que se presenta en 5% al 15% de la población de hipertensos y el déficit de 11β-hidroxiesteroide deshidrogenasa tipo 2 (11β-HSD2). Además, existen causas infrecuentes como el síndrome de Liddle y las formas hipertensivas de hiperplasia suprarrenal congénita (HSC) que son más comunes de ser diagnosticadas en población pediátrica.
Hiperaldosteronismo primario (HAP)El HAP es una de las causas conocidas de hipertensión arterial secundaria. En estos casos la HTA es secundaria a una excesiva y autónoma producción de aldosterona, que a nivel renal induce un aumento en la reabsorción de sal y agua, lo que se traduce en un aumento del volumen intravascular y secundariamente en elevación de la presión arterial (1). Tradicionalmente la prevalencia del HAP ha sido estimada en menos del 1% de los hipertensos cuando la hipokalemia es usada como test de screening (2). Sin embargo, estudios recientes han mostrado que el HAP puede ser mucho más prevalente cuando se miden aldosterona plasmática (AP), la actividad de renina plasmática (ARP) y la relación AP/ARP (ARR) en el screening de esta enfermedad. En el presente milenio, múltiples estudios han usado la determinación de la relación ARR como screening de HAP y han usado el test de supresión con fludrocortisona (TSF) o sobrecarga salina (TSS) para confirmar el diagnóstico (3). Los resultados de estos estudios demuestran que la prevalencia de HAP alcanza cifras cercanas al 5-20% de la población de hipertensos (4). Estas cifras son más altas cuando se consideran hipertensos más severos (estados 2 y 3 del JNC-VI, the Sixth Report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure), donde las cifras pueden elevarse hasta el 15% y en pacientes refractarios a terapia antihipertensiva donde la prevalencia puede llegar hasta el 20% de la población estudiada (5). Además, estos estudios han mostrado que la minoría de los casos presenta hipokalemia, por lo que se ha acuñado el término de HAP normokalémico para identificar esta entidad. Estas evidencias han llevado a plantear que el HAP sería una condición patológica continua en la cual solo una minoría de los sujetos afectados presentaría el cuadro clínico clásico con hipokalemia (6, 7).
La importancia de diagnosticar el HAP, ha tomado relevancia en los últimos años no sólo por su alta prevalencia sino también por los efectos deletéreos de la aldosterona en varios órganos, como ocurre en el corazón y vasos sanguíneos vía receptores no epiteliales e independientes de los cambios en la presión arterial. Este efecto deletéreo en el sistema cardiovascular incluye fibrosis miocárdica, reducción de la fibrinolisis y disfunción endotelial. El estudio RALES (Randomized Aldactone Evaluation Study) y EPHESUS (Eplerenone Post–acute Myocardial Infarction Heart Failure Efficacy and Survival Study) demostraron que la adición de un antagonista de aldosterona a la terapia antihipertensiva, reducía la mortalidad en 30% en pacientes con falla cardiaca tipo IV y reducía la mortalidad en 15% en pacientes con disfunción ventricular izquierda después de un infarto agudo al miocardio (IAM), respectivamente (8, 9). Por su parte, los pacientes con HAP tienen mayor riesgo de eventos cardiovasculares que los esperados para sus niveles de presión arterial, mayor hipertrofia ventricular izquierda, accidente vascular encefálico, e IAM que los pacientes hipertensos esenciales (10). En pacientes con HAP se han observado hallazgos similares, en particular, una patología vascular necrotizante con una marcada infiltración leucocitaria inflamatoria perivascular.
En relación a la etiología del HAP, los subtipos más prevalentes son la hiperplasia adrenocortical bilateral o hiperaldosteronismo idiopático (HI), el adenoma productor de aldosterona (APA) y la hiperplasia adrenal primaria (HP), cuya prevalencia es cercana el 65%, el 35% y el 1% de los HAP, respectivamente. Otras causas son el hiperaldosteronismo familiar tipo I (HF-I) (aldosteronismo remediable por glucocorticoides) y el HF-II (aldosteronismo no remediable por glucocorticoides) y el HF-III.
Enfoque diagnóstico del HAPEl diagnóstico de hiperaldosteronismo primario está destinado a confirmar la autonomía de la secreción de aldosterona del eje renina angiotensina y para ello se utilizan test de screening y confirmatorios.
1Test de screening del HAPLa hipokalemia ha sido considerada como el elemento clásico para el diagnóstico de HAP. Sin embargo, estudios recientes han demostrado que niveles disminuidos de potasio plasmático se presentan solo en 20% de los pacientes afectados por un HAP (11). El potasio plasmático puede ser influenciado por la severidad y duración del hiperaldosteronismo, la ingesta de sodio y la sensibilidad de los túbulos renales a la aldosterona. De esta forma, el HAP normokalémico constituye la forma más común de presentación de la enfermedad, y la variante hipokalémica probablemente la forma más severa de la enfermedad.
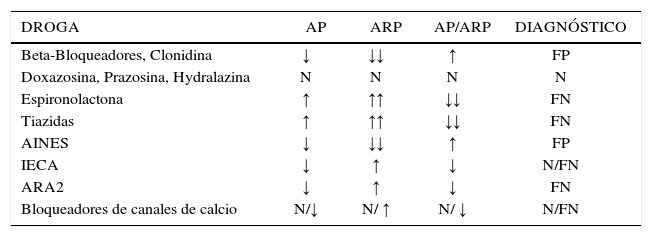
La determinación aislada AP o ARP no es suficiente para el diagnóstico de HAP, en ambos casos hay condiciones que pueden inducir a errores en la determinación. Este margen de error puede ser disminuido si se calcula la relación ARR. Una ventaja de la determinación de la relación es que se ve menos afectada por la administración de drogas y por la posición de los sujetos, permitiendo su uso bajos condiciones menos restrictivas (12). A pesar de esto, debe tenerse presente qué drogas como los diuréticos y los bloqueadores del receptor de angiotensina pueden dar falsos negativos y el propanolol falsos positivos (Tabla 1). Las muestras de AP y ARP deben ser tomadas entre las 8 y las 10 AM después de que el paciente ha permanecido sentado por 15 minutos, y no necesariamente después de estar acostado por 2 horas como indi-caba el protocolo original (13). Los valores de la ARR más usados son entre 20 y 40 cuando la AP es determinada en ng/dl y la ARP ng/ml/h y la conversión con otras unidades ha sido previamente comunicada (14). Recientemente nuestro grupo estudió niños y estableció que la ARR es más baja que en adultos, estableciendo un punto de corte para ARR de 10 (15).
Efecto de drogas antihipertensivas en los niveles de aldosterona y actividad renina plasmática
| DROGA | AP | ARP | AP/ARP | DIAGNÓSTICO |
|---|---|---|---|---|
| Beta-Bloqueadores, Clonidina | ↓ | ↓↓ | ↑ | FP |
| Doxazosina, Prazosina, Hydralazina | N | N | N | N |
| Espironolactona | ↑ | ↑↑ | ↓↓ | FN |
| Tiazidas | ↑ | ↑↑ | ↓↓ | FN |
| AINES | ↓ | ↓↓ | ↑ | FP |
| IECA | ↓ | ↑ | ↓ | N/FN |
| ARA2 | ↓ | ↑ | ↓ | FN |
| Bloqueadores de canales de calcio | N/↓ | N/ ↑ | N/ ↓ | N/FN |
AP: aldosterona plasmática; ARP: actividad renina plasmática; AINES: anti-inflamatorio no esteroidal; IECA: inhibidores de enzima convertidora de angiotesinógeno; ARA2: bloqueadores receptor de angiotensina. FN: falso negativo; FP: falso positivo; N: normal. (Tomado de referencia 14).
La confirmación del diagnóstico de HAP se basa en demostrar la autonomía de la producción de aldosterona a través de un test de supresión. Los más usados son:
a. test de fludrocortisona: los niveles de AP son medidos en la condición basal y después de 4 días de administrar acetato de fludrocortisona (0,4 mg/día, 0,1 mg c/6h) bajo una dieta suplementada con sodio, 110 mmol/día. Las muestras de sangre se toman al quinto día a las 08:00 AM. El test de fludrocortisona es considerado positivo cuando la AP mantiene valores sobre 5 ng/dl (16).
b. test de sobrecarga o infusión salina: consiste en la administración de una solución salina isotónica de 500 ml/hora durante dos a cuatro horas. La persistencia de niveles de AP sobre 5 ng/dl confirma en diagnóstico de HAP (se mide AP a las 9 AM previo a la infusión y al termino de ella). Los autores recomiendan que ambos test sean cuidadosamente monitorizados en ancianos y evitados en pacientes con hipertensión severa, insuficiencia cardiaca severa, accidente vascular, o infarto de miocardio.
c. Prueba de captopril: se efectúa en posición sentada por al menos 60 min antes y durante toda la prueba. A las 9 AM se administra 25 mg de captopril oral y a los 120 min se mide AP y actividad de renina plasmática. El test es positivo si al final la ARR es >30 ó la AP es >8.5 ng/dl. Esta prueba está más indicada en pacientes con riesgo de sobrecarga de volumen (17).
Estudios de Localización del HAPLos procedimientos de localización deben ser llevados a cabo solo después que el diagnóstico de HAP ha sido establecido y son la Tomografía Axial Computada (TAC) y el muestreo de venas suprarrenales.
1. La TAC es un procedimiento muy útil para detectar la mayoría de los adenomas, aún cuando estos sean menores de 5 mm. En pacientes con HI las glándulas suprarrenales aparecen crecidas en forma bilateral o pueden también aparecer de tamaño normal. Aunque no hay una medición exacta del tamaño de la glándula suprarrenal, la TAC es considerada anormal cuando cualquier área de ella es mayor de 10 mm (18). En cambio, los adenomas aparecen como una masa unilateral de baja densidad, generalmente menores de 2 cm. de diámetro. La detección de un nódulo mayor de 6 cm. podría hacer sospechar la presencia de un carcinoma suprarrenal (19). La más clara desventaja de la TAC es cuando la bioquímica de HAP no es claramente definida y la presencia de un incidentaloma podría ser erróneamente confundida con un adenoma. Más aún, una hiperplasia micro-macro nodular con un nódulo dominante podría llevar a un falso diagnóstico de adenoma. La experiencia con resonancia nuclear magnética (RNM) no parece ofrecer ventajas sobre la TAC.
2. Muestreo de venas suprarrenales. Es considerado el método más confiable para probar lateralización como ocurre en los casos de adenoma ó H P. El procedimiento se realiza por vía femoral y se cateterizan ambas venas suprarrenales y la vena cava inferior. Se considera cateterización exitosa si el cortisol es 2 veces mayor en la vena suprarrenal respecto a la vena cava que sirve de control. Se considera que existe lateralización positiva cuando la razón aldosterona/cortisol es > 4 veces a la observada en la vena suprarrenal contralateral. Este procedimiento se realiza bajo infusión de ACTH. Este método requiere considerable experiencia del radiólogo y tiene riesgo de hemorragia suprarrenal. Se recomienda especialmente en sujetos mayores de 40 años en presencia de nódulos suprarrenales, ya que es muy frecuente observar incidentalomas que pueden determinar un falso positivo al estudio de imágenes.
Diagnóstico de HAP FamiliarExisten 3 formas de HAP familiar el tipo 1 (HF-I), el tipo 2 (HF-II) y recientemente ha sido descrito el tipo 3 (HF-III), los cuales difieren en sus formas de presentación y bases genético moleculares.
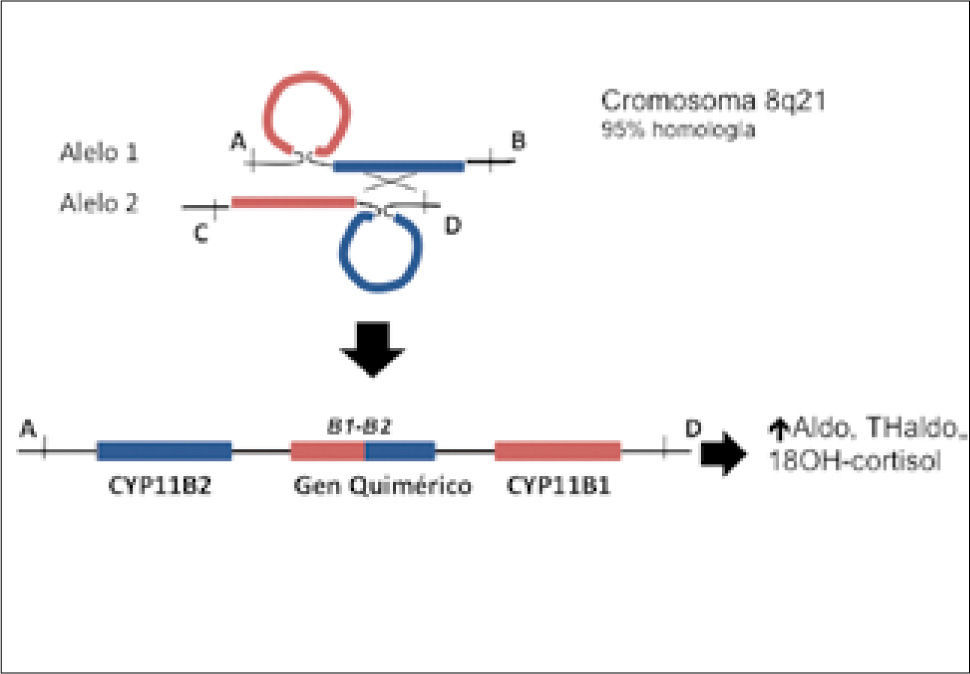
1.-Diagnóstico del HF-IEl HF-I es causado por una recombinación desigual entre los genes que codifica para la 11β-hidroxilasa (CYP11B1) y la aldosterona sintasa (CYP11B2), resultando en un gen quimérico el cual tiene actividad aldosterona sintasa pero es regulado por ACTH (20, 21). Este gen quimérico contiene en su porción amino terminal 3’ los elementos que determinan la respuesta a ACTH fusionado a las secuencias codificadoras del gen CYP11B2 (Figura 1). Este gen es expresado en la zona fasciculata y determina la sobre producción de aldosterona y de los esteroides adrenales 18-hidroxicortisol y 18-oxocortisol, los cuales se encuentran bajo control de ACTH y por tanto son supresibles con glucocorticoides. En la actualidad, es posible detectar la presencia del gen quimérico CYP11B1/CYP11B2 usando la técnica de long-extension PCR, también llamado XL-PCR (1, 22, 23). En pacientes con HF-I la elevación de 18-hidroxicortisol puede ser 4-10 veces el valor normal, en cambio en APA la elevación es discreta y en HI normales (24). Recientemente nuestro grupo estudió niños hipertensos y encontró una prevalencia del 4% de HF-I (25). En la actualidad este test genético se encuentra disponible en la Red de Salud UC.
 y cortisol (CYP11B1) genera un gen quimérico en cromosoma 8, con secuencias reguladoras pertene-cientes a CYP11B1 y secuencias codificantes de CYP11B2. La localización del gen quimérico en la zona fasciculata en la corteza de la glándula suprarrenal, permite una elevada síntesis de aldosterona dado que se encuentra regulado por ACTH y no por angiotensina II.")
Gen quimérico CYP11B1/CYP11B2. La recombinación desigual entre el gen encargado de la síntesis de aldosterona (CY11B2) y cortisol (CYP11B1) genera un gen quimérico en cromosoma 8, con secuencias reguladoras pertene-cientes a CYP11B1 y secuencias codificantes de CYP11B2. La localización del gen quimérico en la zona fasciculata en la corteza de la glándula suprarrenal, permite una elevada síntesis de aldosterona dado que se encuentra regulado por ACTH y no por angiotensina II.
Esta forma de presentación es clínicamente indistinguible de HAP esporádico, respecto a la edad de diagnóstico, género, frecuencia de hipokalemia, A P, ARP (26). El HF-II puede presentarse como una hiperplasia adrenocortical bilateral o como un adenoma. El modo de herencia del HF-II se mantiene especulativo, pero la transmisión vertical sugiere una herencia dominante autosómica. Se especula que el gen responsable estaría localizado en el cromosoma 7 (cr 7p22) asociado a HF-II.
3.-Diagnóstico de HF-IIIEs una forma autosómica dominante de hiperaldosteronismo causada por una mutación en el gen KCNJ5, el cual codifica para un canal selectivo para el ion potasio. Este cuadro se caracteriza por niveles elevados de aldosterona, ARR y 18-hidroxicortisol (27). Al ocurrir la mutación se produce un aumento de la conductancia al sodio que induce la despolarización de la membrana, lo que induce secundariamente un aumento de la síntesis de aldosterona y proliferación de las células de la glomerulosa. Sin embargo, publicaciones recientes han demostrado que APA del tipo esporádicos presentan entre un 14 al 65% de mutaciones en el gen KCNJ5. Más aún, todas las mutaciones comunicadas corresponden a la G151R (sustituye arginina por glicina en el aminoácido 151) o L186R (sustituye arginina por leucina en el aminoácido 186) (28-30).
Terapia del HAPEl objetivo del tratamiento es prevenir la morbi-mortalidad asociada con la hipertensión, las alteraciones hidroelectrolíticas y el daño cardiovascular. La aproximación terapéutica del HAP depende del subtipo etiológico. Así, la cirugía es el tratamiento de elección en pacientes con APA e hiperplasia adrenal primaria. En la actualidad la cirugía laparoscópica es de elección dado que presenta menos complicaciones y los períodos de hospitalización y recuperación son más cortos. La corrección quirúrgica del HAP generalmente normaliza o disminuye significativamente las cifras de presión arterial (31). La persistencia de la hipertensión puede estar relacionada con la severidad o cronicidad de la enfermedad hipertensiva o por la coexistencia de hipertensión esencial.
El tratamiento farmacológico es la terapia de elección para pacientes afectados por HI. La espironolactona, un antagonista de aldosterona a nivel de su receptor, ha sido la droga tradicionalmente usada. Las dosis varían entre 25-100 mg/día, con lo cual se alcanza un efectivo control de la presión arterial y de la hipokalemia en la mayoría de los casos. Sin embargo, su uso produce efectos adversos como ginecomastia, disfunción eréctil, disminución de la libido, síntomas gastrointestinales e irregularidades menstruales. La eplerenona es un nuevo antagonista selectivo del receptor de mineralocorticoides recientemente aprobado para el tratamiento de la hipertensión arterial. La ventaja con respecto a la espironolactona es que la eplerenona no presenta los efectos adversos descritos para la espironolactona y por ende aparece como una droga de elección para el manejo de estos pacientes. Otras alternativas de tratamiento son el amiloride y el triamterene, drogas que impiden la acción de aldosterona al inducir un bloqueo del canal de sodio a nivel renal y con ello impiden la retención de sodio y la pérdida de potasio (32). El nifedipino, un bloqueador de los canales de calcio, también ha mostrado ser efectivo en el control de la presión arterial, pero los resultados a largo plazo han sido poco alentadores. En los pacientes con un HF-I el tratamiento de elección es la dexametasona a bajas dosis entre 0.125–0.5 mg/día (33). Sin embargo, también pueden responder a prednisona o hidrocortisona. En niños es recomendable ajustar la dosis por superficie corporal, para evitar una sobre dosificación.
Otras formas de hipertensión arterial mineralocorticoide1Déficit de 11β-hidroxiesteroide deshidrogenasa tipo 2 (11β-HSD2)La regulación del cortisol es importante para controlar la presión arterial. A nivel celular, la concentración de cortisol está regulada por la actividad de las enzimas 11β-HSD2 que inactiva el cortisol en cortisona y 11β-hidroxiesteroide deshidrogenasa tipo 1 (11β-HSD1) que convierte la cortisona inactiva a cortisol (34).
En condiciones normales, la 11β-HSD-2 inactiva el cortisol a cortisona principalmente en los tejidos donde se expresa el receptor de mineralocorticoides, como el riñón y el colon (Figura 2). Esta inactivación tiene la función de proteger al receptor mineralocorticoideo (MR) del exceso de cortisol en estos tejidos, dado que esta hormona circula en mayor concentración que la aldosterona y tiene gran afinidad por el MR lo que produciría una activación del receptor, aumentando la reabsorción de agua y sal, con la consecuente expansión de volumen e hipertensión (35).
. La enzima 11β-HSD2 es la encargada de inactivar el cortisol (F) en cortisona (E) y con ello impedir que active el receptor de mineralocorticoides. Cuando existe un déficit de esta enzima, el cortisol no es inactivado y puede unirse libremente al receptor de mineralocorticoides, activándolo.")
Actividad de 11β-hidroxiesteroide deshidrogenasa tipo 2 (11β-HSD2). La enzima 11β-HSD2 es la encargada de inactivar el cortisol (F) en cortisona (E) y con ello impedir que active el receptor de mineralocorticoides. Cuando existe un déficit de esta enzima, el cortisol no es inactivado y puede unirse libremente al receptor de mineralocorticoides, activándolo.
Los defectos congénitos o adquiridos en la enzima 11β-HSD2 resultan en un exceso de actividad mineralocorticoide cortisol-dependiente. El déficit congénito autosómico recesivo fue descrito por primera vez por Ulick en 1979 quien acuño el término de síndrome de exceso aparente de mineralocorticoides (SEAM) (36). Su presentación clínica clásica incluye: bajo peso al nacer, retraso del crecimiento, talla baja, hipertensión severa, hipokalemia, supresión en la ARP y aldosterona indetectable (37).
Existe una creciente evidencia de que un déficit parcial en la actividad de la 11β-HSD2, ya sea por mutaciones, polimorfismos e inhibidores endógenos ó exógenos, pueden desempeñar un papel en el desencadenamiento de la hipertensión. Su actividad se puede estudiar mediante la relación cortisol /cortisona en suero (8-10 AM) ó en orina de 24 hrs (34). En una reciente publicación de nuestro grupo, observamos un déficit parcial de la actividad de la enzima 11β-HSD2 en el 15,7% de los pacientes hipertensos esenciales estudiados (37). Estos nuevos hallazgos son de gran importancia para el manejo, ya que los pacientes afectados por un déficit parcial de 11β-HSD2 podrían ser tratados con el bloqueo específico del receptor mineralocorticoide y/o con corticoides de acción prolongada sin actividad mineralocorticoide como dexametasona o betametasona.
2Síndrome de LiddleEste síndrome es causado por mutaciones en el canal epitelial renal de sodio (ENaC). Este canal es considerado el paso limitante para la absorción de sodio en el túbulo distal y está compuesto por 3 subunidades denominadas alfa, beta y gama. En el síndrome de Liddle se han encontrado mutaciones en las subunidades beta y gama. Como resultado de estas mutaciones se produce una activación constitutiva del canal que lleva a un aumento de la reabsorción de sodio y agua y secundariamente a la expansión del volumen intravascular. Este desorden es heredado en forma autosómica dominante y los pacientes afectados presentan hipertensión y niveles suprimidos de renina y aldosterona. Este desorden responde a inhibidores del transporte epitelial de sodio como es el triamterene, pero no a antagonistas del receptor de mineralocorticoides como es la espironolactona. Los pacientes afec-tados también responden al trasplante renal, lo cual resulta en normalización de la presión arterial y de las alteraciones hidroelectrolíticas.
3Formas hipertensivas de hiperplasia suprarrenal congénitaLa deficiencia de 11β-hidroxilasa es la causa más común y está presente en el 5% de todos los casos. Este desorden es causado por mutaciones en el gen CYP11B1, que codifica para la enzima 11β-hidroxilasa el cual es el encargado de convertir el deoxicortisol a cortisol. Como consecuencia de la deficiente producción de cortisol, la concentración de ACTH se eleva, estimulando la esteroidogénesis suprarrenal y con ello la elevación de los niveles plasmáticos de deoxicorticosterona. Dado que la deoxicorticosterona tiene actividad mineralocorticoidea, su exceso causa retención de sal y agua, supresión de renina y aldosterona e hipertensión arterial. Este desorden responde a dosis supresoras de dexametasona. La deficiencia de 17á-hidroxilasa es una forma rara de hiperplasia suprarrenal causada por defectos en el citocromo P450c17, enzima que tiene actividad de 17á-hidroxilasa y 17,20-liasa. Este desorden es caracterizado por la ausencia de la síntesis de esteroides sexuales y por una disminución de la síntesis de cortisol con hipersecreción compensatoria de ACTH. La ACTH estimula la síntesis de grandes cantidades de deoxicorticosterona y corticosterona, lo cual determina la aparición de hipertensión arterial. Las bases moleculares de este defecto han sido clarificadas en al menos dos docenas de pacientes, identificando al menos 17 diferentes lesiones en el gen CYP17 que codifica para el citocromo P450c17. Los pacientes afectados pueden ser tratados con dosis supresoras de dexametasona.
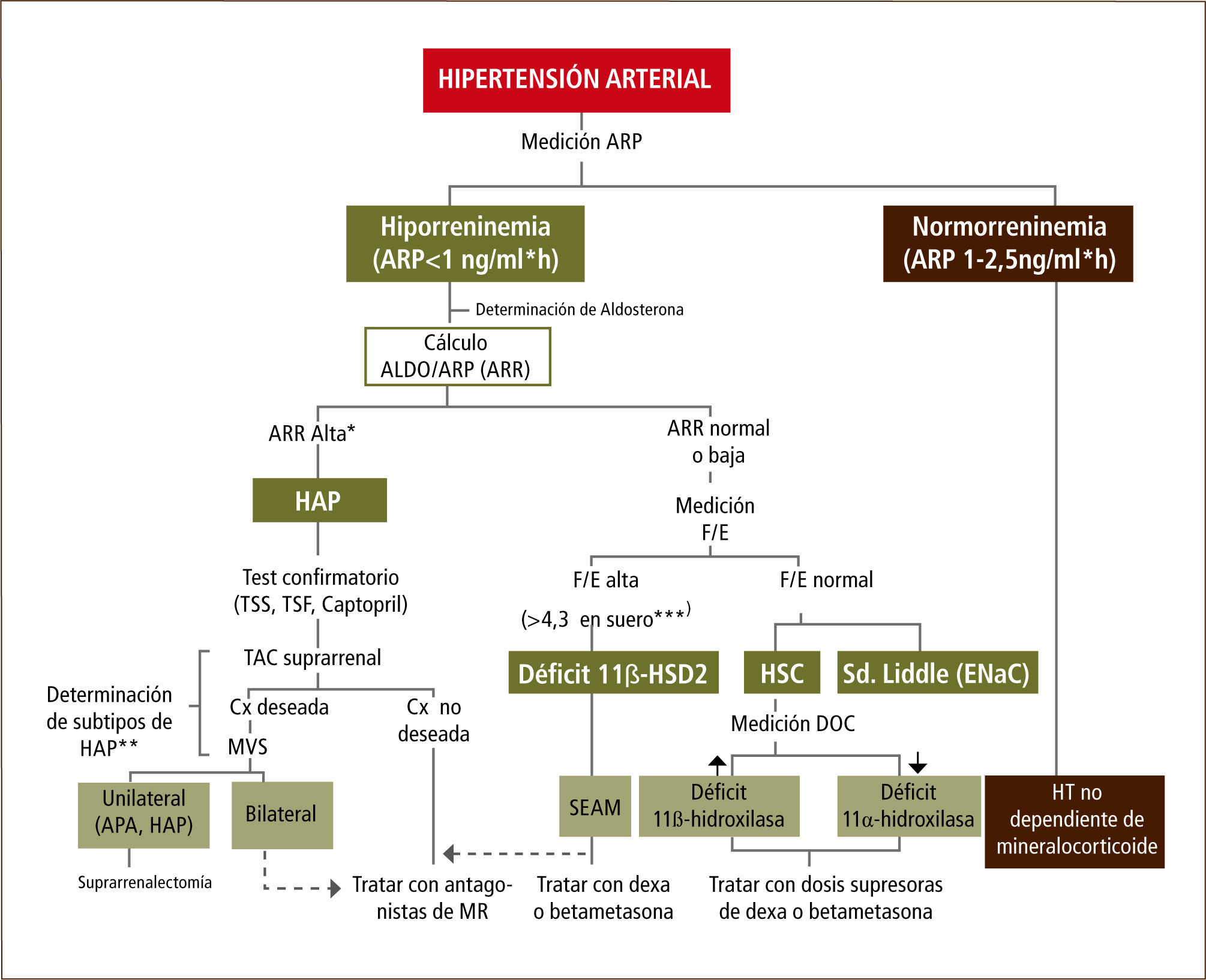
En resumen en la Figura 3 se muestra un algoritmo para la identificación de los distintos sub-tipos de hipertensión arterial mineralocorticoidea.
 y ≥10 en niños sugiere HAP 15 (15). ** Se sugiere el análisis genético de gen quimérico (HF-I) (14, 22). Para aquellos pacientes que son gen quimérico (GQ) positivo se recomienda la terapia con bajas dosis de betametasona. Si los niveles de ARR son muy elevados y GQ (-), se sugiere la presencia de HF-II o HF-III, para este último realizar un estudio genético para el gen KCNJ528. *** El punto de corte para la razón F/E corresponde a niños normotensos, valor que puede aumentar con la edad en sujetos normotensos (38).")
Algoritmo para la detección, confirmación y análisis de subtipos de hipertensión arterial mineralocorticoidea
Abreviaciones: ARP: actividad renina plasmática; ARR: razón aldosterona/ARP; HAP: hiperaldosteronismo primario; F: cortisol; E: cortisona; TSS: test de sobrecarga salina; TSF: test de supresión con fludrocortisona; TAC: tomografía axial computarizada; HSC: hiperplasia suprarrenal congénita; Cx: cirugía; MVS: muestras de venas suprarrenales; APA: adenoma productor de aldosterona; MR: receptor mineralocorticoideo; DOC: deoxicorticosterona; SEAM: Síndrome de exceso aparente de mineralocorticoides; HE: hipertensión esencial. * Se considera la razón ARR ≥ 25 en adultos (5) y ≥10 en niños sugiere HAP 15 (15). ** Se sugiere el análisis genético de gen quimérico (HF-I) (14, 22). Para aquellos pacientes que son gen quimérico (GQ) positivo se recomienda la terapia con bajas dosis de betametasona. Si los niveles de ARR son muy elevados y GQ (-), se sugiere la presencia de HF-II o HF-III, para este último realizar un estudio genético para el gen KCNJ528. *** El punto de corte para la razón F/E corresponde a niños normotensos, valor que puede aumentar con la edad en sujetos normotensos (38).
Los autores declaran no tener conflictos de interés, relacionados a este artículo.