




Los síndromes poliglandulares autoinmunes comprenden un amplio espectro de trastornos endocrinos. Estos síndromes incluyen trastornos monogénicos como el síndrome poliglandular tipo 1 (tipo juvenil) y trastornos genéticos complejos como el síndrome poliglandular tipo 2 (tipo adulto). Estos trastornos se basan en la presencia de infiltración linfocitaria de la glándula afectada, anticuerpos órgano-específico en sangre, defectos en la inmunidad celular y asociación con los genes de los antígenos leucocitarios humanos (HLA) o con genes de respuesta inmune.
Cualquier sistema endocrino puede ser afectado, pero ocurre en ciertos patrones, por ejemplo, Tiroiditis de Hashimoto con diabetes tipo 1 y Enfermedad de Addison.
Es muy importante la sospecha y detección precoz de esta asociación de enfermedades, dado que, de esta forma, podemos evitar morbilidad e incluso mortalidad en nuestros pacientes.
The polyglandular autoimmune syndromes comprise a wide spectrum of autoinmune disorders. These syndromes include monogenic disorders such as polyglandular autoinmune syndrome type 1 (juvenile type) and complex genetic disorders such as polyglandular autoinmune syndrome type 2 (adult type). These disorders are based on the presence of lymphocyte infiltration in the affected gland, organ-specific antibodies in serum, celular immune defects and association with the human leucocyte antigen (HLA) genes or immune response genes.
Any endocrine gland can be affected but in some patterns, for example, Hashimoto’s thyroiditis with type 1 diabetes and Addison’ s disease. Is very important an early suspicion and detection of these associated disorders, because we can prevent morbidity and even mortality in our patients.
La autoinmunidad puede afectar a cualquier glándula del sistema endocrino. Modelos animales y estudios en humanos destacan la importancia de los alelos HLA (human leucocyte antigen) como moléculas desencadenantes de autoinmunidad específica contra los tejidos, provocando una pérdida de la propia tolerancia.
Enfermedades como la Diabetes tipo 1 A, Tiroiditis de Hashimoto, Enfermedad de Addison, Enfermedad de Graves, entre otras, son el resultados de una destrucción tisular mediada por mecanismos autoinmunes (1). Cada uno de estos trastornos presenta una evolución natural, la que se puede dividir en etapas, comenzando con una susceptibilidad genética, desencadenantes ambientales, autoinmunidad activa y finalmente trastorno de la función normal con enfermedad clínica.
Las enfermedades autoinmunes afectan al 5 a 10% de la población y un gran porcentaje de éstas compromete una glándula endocrina. Cualquier glándula endocrina puede ser objeto del sistema inmune y frecuentemente pueden ser múltiples los órganos comprometidos en un mismo individuo, pudiendo conformar síndromes poliglandulares autoinmunes (2).
La historia familiar de autoinmunidad y el estudio de los autoanticuerpos puede identificar a aquellos individuos en riesgo.
El conocimiento de estas enfermedades y la relación entre ellas puede llevarnos a un diagnóstico más temprano y con ello a una menor morbilidad y mortalidad.
A pesar del progreso en el entendimiento de estas enfermedades, la terapia de reemplazo hormonal sigue siendo el pilar del tratamiento.
Historia de autoinmunidad en órganos endocrinosEn 1855 Thomas Addison describió por primera vez los signos y síntomas de “un estado caracterizado por anemia, debilidad, pulso débil, irritabilidad estomacal y un cambio peculiar en el color de la piel que ocurre en relación a un trastorno de la cápsula suprarrenal” (3).
En las autopsias de sus pacientes encontró en orden decreciente las siguientes causas: tuberculosis, neoplasia, hemorragia adrenal y un caso de fibrosis suprarrenal de origen desconocido, la que describió como ocasionada por una inflamación que había destruido la glándula hasta su atrofia. Esta fue la primera descripción de adrenalitis autoinmune en la literatura. El Dr. Addison observó además que el paciente presentaba lesiones en la piel compatibles con lo que hoy llamamos vitíligo. Posteriormente el vitíligo ha sido reconocido como una enfermedad autoinmune que nos debe hacer sospechar de otras patologías inmunológicas en nuestros pacientes.
En 1926 Schmidt describió dos pacientes que presentaban asociación de Enfermedad de Addison no tuberculosa con tiroiditis linfocítica crónica llamada posteriormente Síndrome de Schmidt.
En 1964 Carpenter et al, reportaron un caso de Síndrome de Schmidt asociado a diabetes mellitus tipo 1.
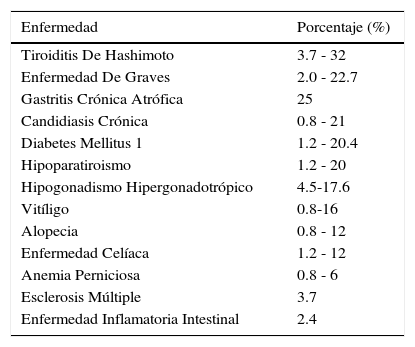
Y así sucesivamente se ha descrito asociación entre patología autoinmune como enfermedad de Addison, enfermedad tiroidea, gastritis crónica atrófica, diabetes mellitus tipo 1, hipoparatiroidismo, hipogonadismo, vitíligo, alopecia, enfermedad celíaca, anemia perniciosa, esclerosis múltiple, enfermedad inflamatoria intestinal, síndrome de Sjögren, hepatitis crónica e hipofisitis linfocitaria (Tabla 1).
Prevalencia de enfermedad autoinmune en población con enfermedad de addison
| Enfermedad | Porcentaje (%) |
|---|---|
| Tiroiditis De Hashimoto | 3.7 - 32 |
| Enfermedad De Graves | 2.0 - 22.7 |
| Gastritis Crónica Atrófica | 25 |
| Candidiasis Crónica | 0.8 - 21 |
| Diabetes Mellitus 1 | 1.2 - 20.4 |
| Hipoparatiroismo | 1.2 - 20 |
| Hipogonadismo Hipergonadotrópico | 4.5-17.6 |
| Vitíligo | 0.8-16 |
| Alopecia | 0.8 - 12 |
| Enfermedad Celíaca | 1.2 - 12 |
| Anemia Perniciosa | 0.8 - 6 |
| Esclerosis Múltiple | 3.7 |
| Enfermedad Inflamatoria Intestinal | 2.4 |
Esta asociación entre patología autoinmune endocrina y no endocrina se ha clasificado en lo que llamamos síndromes poliglandulares, los cuales presentan de base, trastornos genéticos muy diferentes entre ellos.
Sindrome poliglandular (SPG) definición y clasificaciónLos síndromes poliglandulares autoinmunes (SPG) se definen como la falla autoinmune de al menos dos glándulas. Comprenden un amplio espectro de trastornos autoinmunes, pero en una asociación que no es al azar, sino en patrones repetidos, lo que llevó en 1980 a Neufeld y Blizzard a clasificar en cuatro tipos a estos síndromes (4) (Tabla 2).
Clasificación original de sindrome poliglandular según los autores neufeld y blizzard
| SPG 1 | Candidiasis Crónica, Hipoparatiroidismo Crónico, Insuficiencia Suprarrenal Autoinmune. (al menos dos presentes). |
| SPG 2 | Insuficiencia Suprarrenal Autoinmune, Enfermedad Tiroidea Autoinmune y/o Diabetes Mellitus 1. (insuficiencia suprarrenal debe estar siempre presente). |
| SPG 3 | Enfermedad Tiroidea Autoinmune y otras enfermedades autoinmunes. (excluye insuficiencia suprarrenal) |
| SPG 4 | Dos o más enfermedades autoinmunes que no clasifiquen en los otros tipos. |
Posteriormente se han realizado modificaciones a esta clasificación inicial, tendiendo a clasificar dentro del SPG 2 al 3 y 4 (7, 8).
Sindrome poliglandular tipo 1 (SPG 1)Se caracteriza por tres componentes principales: candidiasis crónica, hipoparatiroidismo e insuficiencia suprarrenal autoinmune. Para clasificar como SPG 1 deben estar presente al menos dos de los componentes principales. En el 50% de los casos, la presentación clínica tiene un orden cronológico, primero aparece la candidiasis, luego el hipoparatiroidismo y la insuficiencia suprarrenal. En el otro 50% se presentan todas juntas.
Su prevalencia es muy baja, y dependiendo de la etnia oscila entre 1/9000 y 1/80000 habitantes.
I.-InmunogenéticaEl SPG 1 es la primera enfermedad autoinmune que ha demostrado ser causada por la mutación de un sólo gen, es decir, es una enfermedad monogénica.
En 1994 se encontró el gen responsable de este síndrome, localizado en el brazo largo del cromosoma 21. Este gen llamado AIRE (autoinmune regulator gene), consiste en 14 exones y codifica para una proteína de 545 aminoácidos, la cual tiene un rol regulador de transcripción nuclear (5).
Esta proteína se expresa en células estromales de los tejidos linfoides, incluyendo a las células epiteliales tímicas. El gen AIRE regula la transcripción de antígenos restringidos a ciertos tejidos y se ha relacionado con la tolerancia inmune central y periférica.
Se han descrito más de 40 mutaciones en el gen AIRE y cuando estas mutaciones están presentes se pierde la tolerancia a múltiples antígenos propios (1).
Los pacientes con SPG 1 tienen autoanticuerpos dirigidos a tejidos específicos y a enzimas clave en su función, tal como en la insuficiencia suprarrenal donde los anticuerpos están dirigidos contra la 21 hidroxilasa, enzima clave en la síntesis de glucocorticoides.
Evidencia reciente sugiere que la autoinmunidad y la inmunodeficiencia pueden estar estrictamente asociadas. Uno de los mecanismos está relacionado con la habilidad disminuida del sistema inmune para eliminar las infecciones en pacientes con inmunodeficiencia lo que causa activación inmunológica perpetua y con esto, autoinmunidad. La autoinmunidad se debe a una combinación de múltiples variedades genéticas, infecciones y factores inmunoreguladores que junto con un fenotipo autoinmune puede incrementar la susceptibilidad a infecciones. En el caso de SPG 1 se evidencia claramente esta combinación (6).
II.-Componentes del Cuadro Clínico de SPG 1IIa.-Candidiasis CrónicaAfecta principalmente uñas, piel, lengua, mucosas y en las comisuras labiales (queilosis angular). Generalmente ocurre antes de los cinco años y es una expresión de deficiencia inmunológica selectiva de las células T a Cándida Albicans, combinada con una respuesta normal de células B frente a los antígenos de Cándida, lo que previene el desarrollo de candidiasis sistémica. Existe el riesgo de desarrollo de carcinoma de la mucosa oral como consecuencia de la candidiasis crónica. Se debe usar terapia antifúngica en estos pacientes, pero no siempre se obtiene buenos resultados, especialmente en cuanto al compromiso de mucosas (3).
IIb.-Hipoparatiroidismo CrónicoDel déficit de hormona paratiroidea resulta la hipocalcemia. Un estudio reciente identificó un autoantígeno paratiroideo, llamado NALP 5 (Familia NACHT, leucine-rich-repeat protein 5) cuyos anticuerpos están presentes sólo en los pacientes con SPG1 que tienen hipoparatiroidismo, no en el resto de los SPG1 ni en otros trastornos endocrinos (5).
El hipoparatiroidismo se desarrolla generalmente después de los 10 años de edad, apareciendo después de la candidiasis. Si aparece en el período neonatal se debe hacer un diagnóstico diferencial, especialmente con el Síndrome de Di George (hipoparatiroidismo y ausencia de timo por defecto de línea media). La presencia de anticuerpos anti paratiroideos o antimitocondriales es muy variable en la literatura.
IIc.-Insuficiencia SuprarrenalGeneralmente aparece después de los 15 años de edad y en la mayoría de los casos existen anticuerpos anticorteza suprarrenal (ACA) y anti 21 hidroxilasa (AC 21OH) desde mucho antes de que se manifieste la clínica.
IId.-Otros trastornos asociadosOtras endocrinopatías, como hipogonadismo hipogonadotrópico, tiroiditis crónica, diabetes mellitus 1, hipofisitis autoinmune.
Enfermedades gastrointesinales autoinmune tales como enfermedad celíaca y anemia perniciosa.
Alopecia, vitíligo, enfermedad reumática, síndrome de Sjögren, distrofia ectodérmica.
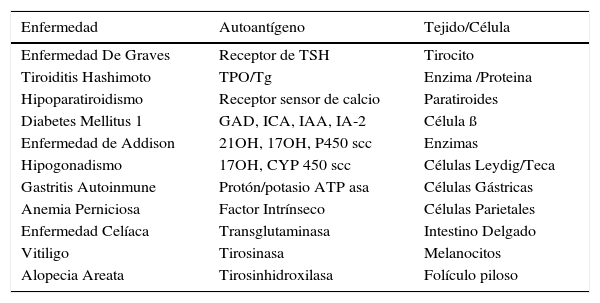
SPG 1 es el síndrome autoinmune que tiene la mayor combinación de enfermedades autoinmunes y de autoanticuerpos en forma simultánea en un mismo individuo (Tabla 3).
Identificación de autoantígenos y tejidos afectados en cada enfermedad autoinmune
| Enfermedad | Autoantígeno | Tejido/Célula |
|---|---|---|
| Enfermedad De Graves | Receptor de TSH | Tirocito |
| Tiroiditis Hashimoto | TPO/Tg | Enzima /Proteina |
| Hipoparatiroidismo | Receptor sensor de calcio | Paratiroides |
| Diabetes Mellitus 1 | GAD, ICA, IAA, IA-2 | Célula ß |
| Enfermedad de Addison | 21OH, 17OH, P450 scc | Enzimas |
| Hipogonadismo | 17OH, CYP 450 scc | Células Leydig/Teca |
| Gastritis Autoinmune | Protón/potasio ATP asa | Células Gástricas |
| Anemia Perniciosa | Factor Intrínseco | Células Parietales |
| Enfermedad Celíaca | Transglutaminasa | Intestino Delgado |
| Vitiligo | Tirosinasa | Melanocitos |
| Alopecia Areata | Tirosinhidroxilasa | Folículo piloso |
El reemplazo hormonal es el pilar del tratamiento en estos pacientes. La candidiasis mucocutánea necesita ser tratada en forma agresiva y se debe monitorear las recurrencias que pueden ocurrir a lo largo de todo el tubo digestivo, ya que en enfermedad no tratada se puede desarrollar cáncer del epitelio.
El hipoparatiroidismo debe ser tratado con suplementación de calcio, magnesio y vitamina D.
Se debe buscar la presencia de asplenia y así administrar vacunas para neumococo, meningococo y haemophilus influenza.
A los pacientes que aun no han presentado enfermedad de Addison, se les debe determinar anticuerpos anti 21 hidroxilasa si se encuentran disponibles, sino debe controlarse cortisol basal y ACTH y/o test de ACTH para cortisol anualmente (1).
En los pacientes que aun no han presentado diabetes tipo 1 se les debe determinar anticuerpos antiislotes, anti GAD y anti insulina (Tabla 3), con seguimiento de glicemia cada tres a seis meses.
El estudio genético del GEN AIRE se puede realizar para confirmar la enfermedad en el paciente con SPG1, pero aun más importante es hacerlo en hermanos o familiares menores que el caso índice.
Sindrome poliglandular tipo 2 (SPG 2)Es una condición rara, pero bastante más frecuente que el SPG1, también llamado Síndrome de Schmidt. Tiene una prevalencia de 1.4 – 2 por 100.000 habitantes. Puede ocurrir a cualquier edad y en ambos sexos, pero es más frecuente que ocurra en mujeres en la edad media de la vida y es muy raro que ocurra en la niñez.
I.-Componentes del cuadro clínicoSe caracteriza por la presencia de Insuficiencia Suprarrenal Autoinmune (Enfermedad de Addison), asociada a enfermedad tiroidea autoinmune y/o diabetes mellitus tipo 1.
La Enfermedad de Addison está presente en el 100% de los casos, la enfermedad tiroidea en el 69 a 82% y la diabetes mellitus tipo 1 en el 30 a 52% de los pacientes (3).
En el inicio de la Enfermedad de Addison los anticuerpos anti corteza suprarrenal y los anticuerpos anti 21 hidroxilasa son detectables en la mayoría de los pacientes.
Los pacientes con tiroiditis crónica presentan frecuentemente anticuerpos anti tiroperoxidasa (ATPO) y anticuerpos antitiroglobulina (ATG) y los pacientes con Enfermedad de Graves presentan anticuerpos anti receptor de TSH (TRAB).
Los pacientes con Diabetes Tipo 1 frecuentemente presentan anticuerpos positivos tales como ICA (anticuerpos anti-islotes), GAD (glutamato decarboxilasa), IAA (anti-insulina) y/o IA2 (anti-tirosina fosfatasa), especialmente al inicio de la enfermedad, incluso varios años antes de presentarla y al evaluar su presencia en los familiares directos, éstos tienen una prevalencia mayor a la de la población general.
En el SPG 2 la diabetes mellitus 1 y la enfermedad de Graves generalmente se presentan antes de la Enfermedad de Addison, en cambio la Tiroiditis Crónica Autoinmune (Enfermedad de Hashimoto) puede presentarse al mismo tiempo o después de la Enfermedad de Addison.
Conociendo la historia natural de SPG 2 y sus diferentes formas de presentación, adquiere importancia el valor de los anticuerpos en estos pacientes, es decir, algunos autores recomiendan que en el debut de una diabetes mellitus 1 se midan los anticuerpos correspondientes a la diabetes más los tiroideos y los ACA y AC 21OH, si están disponibles. Este enfrentamiento podría identificar precozmente a los pacientes susceptibles de presentar un SPG 2, lo cual podría reducir morbilidad e incluso mortalidad, al detectar precozmente una posible insuficiencia suprarrenal en el curso de la enfermedad.
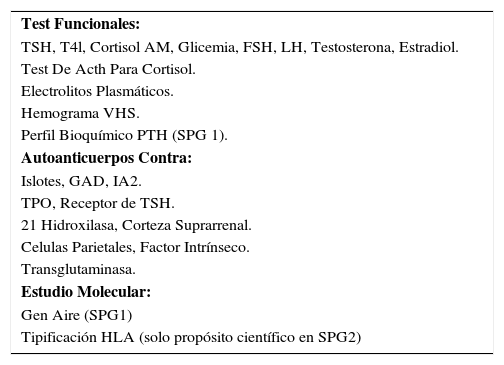
Se debiera considerar como “la punta del iceberg” a los pacientes que presentan una enfermedad autoinmune que conforma un SPG, en este sentido, el estudio funcional en forma regular de las otras patologías también debe recomendarse, ya que muchas veces se las podrá encontrar en estado subclínico y su enfrentamiento será precoz, y con menor morbilidad que en casos donde no se les ha sospechado previamente. Así, se recomienda realizar estudio de TSH, T4 libre, test de ACTH y curva de glucosa, obviando el estudio que no corresponda cuando la enfermedad ya esté presente. Esto también se recomienda en los otros tipos de SPG, como por ejemplo, solicitar PTH y perfil bioquímico en patologías con sospecha de SPG 1 (Tabla 4).
Screening para estudio de spg
| Test Funcionales: |
| TSH, T4l, Cortisol AM, Glicemia, FSH, LH, Testosterona, Estradiol. |
| Test De Acth Para Cortisol. |
| Electrolitos Plasmáticos. |
| Hemograma VHS. |
| Perfil Bioquímico PTH (SPG 1). |
| Autoanticuerpos Contra: |
| Islotes, GAD, IA2. |
| TPO, Receptor de TSH. |
| 21 Hidroxilasa, Corteza Suprarrenal. |
| Celulas Parietales, Factor Intrínseco. |
| Transglutaminasa. |
| Estudio Molecular: |
| Gen Aire (SPG1) |
| Tipificación HLA (solo propósito científico en SPG2) |
Otras enfermedades autoinmunes pueden estar presentes en SPG 2 con sus respectivos autoanticuerpos incluso precediéndolas, aunque con menor frecuencia que en SPG 1, tales como vitíligo, alopecia, hipogonadismo hipergonadotrófico, hipofisitis autoinmune, miastenia gravis, gastritis crónica atrófica con o sin anemia perniciosa y hepatitis autoinmune.
II.-PatogeniaModelos animales sobre la patogenia del SPG 2 son compatibles con la teoría de una infección viral que desencadena una enfermedad autoinmune.
El llamado “mimetismo molecular” se caracteriza por una respuesta inmune a un agente del ambiente que tiene una reacción cruzada con un antígeno del huésped, lo que resulta en enfermedad (7).
También se la ha atribuido un rol a una desregulación de la vía apoptótica. Una disfunción de la vía apoptótica Fas o la producción de factores solubles incluyendo Fas solubles y Fas ligandos, puede tener relación con la patogenia de la enfermedad endocrina autoinmune. En el caso de la diabetes mellitus tipo 1 se ha postulado una susceptibilidad aumentada de las células â de los islotes de Langerhans a la inducción de apoptosis por linfocitos T citotóxicos, presumiblemente a través de estos receptores de superficie de la vía Fas, lo que podría ser responsable de una muerte facilitada de las células â (7).
III.-InmunogenéticaEl SPG 2 generalmente se presenta en varias generaciones de una misma familia.
Es una enfermedad autosómica dominante, pero con penetrancia incompleta, es decir, se ha encontrado una frecuencia aumentada de enfermedad autoinmunes en familiares de primer grado de pacientes con SPG 2. Es una enfermedad poligénica, no como el SPG1 cuya alteración está en un sólo gen, es decir, es una enfermedad monogénica.
De los modelos animales en enfermedad autoinmune, se ha podido deducir que los genes que se ocupan del complejo mayor de histocompatibilidad, en particular los de respuesta inmune tales como los HLA-DQ Y HLA DR, son esenciales en gatillar la enfermedad (8). Dentro de estos destacan los genes del antígeno leucocitario humano (HLA) en el cromosoma 6 y el gen del antígeno linfocitario citotóxico T (CTLA-4) en el cromosoma 2 (2q33) que comprende 4 exones y codifica para una proteína que actúa como moduladora de la activación de la activación de células T.
Muchos SPG 2 se asocian con una frecuencia aumentada de los haplotipos HLA A1, B8, DR3, DQA1*0501, DQB1*0201. La Enfermedad de Addison se asocia fuertemente a DR3 y DR4 (7).
Dado que es multifactorial no se recomienda realizar estudio genético en SPG 2, salvo que sea con fines científicos.
Síndrome poliglandular tipo 3Corresponde a la asociación entre enfermedad autoinmune tiroidea y otra enfermedad autoinmune donde no se encuentre la Enfermedad de Addison.
Se ha descrito asociación entre enfermedad autoinmune tiroidea como Tiroiditis de Hashimoto, mixedema idiopático, Enfermedad de Graves y Oftalmopatía de Graves con Síndrome de Hirata, Diabetes mellitus tipo 1, hipofisitis linfocitaria, falla ovárica prematura, gastritis crónica atrófica con o sin anemia perniciosa, enfermedad celíaca, enfermedad inflamatoria intestinal, hepatitis autoinmune, cirrosis biliar primaria, vitíligo, alopecia, miastenia gravis, esclerosis múltiple, lupus, artritis reumatoide, síndrome de Sjögren, vasculitis, síndrome antifosfolípidos, esclerosis sistémica entre otros.
Síndrome poliglandular tipo 4Se refiere a la combinación de dos o más enfermedades autoinmunes que no clasifiquen en las categorías anteriores. Por ejemplo, Enfermedad de Addison más hipogonadismo.
Tratamiento del sindrome poliglandular tipo 2 (SPG3 y SPG4)En el presente, el tratamiento del síndrome poliendocrino está condicionado por el manejo de cada trastorno individual, es decir, por la sustitución de cada hormona deficitaria.
Una de las consideraciones más importantes en este tema es la siguiente: Frente a un paciente con hipotiroidismo debe sospecharse la concomitancia con enfermedad de Addison, dado que el inicio de la terapia con levotiroxina puede precipitar una crisis addisoniana. De confirmarse la insuficiencia suprarrenal, el tratamiento con glucocorticoides debe preceder a la administración de levotiroxina. Esto se debe a que la sustitución con levotiroxina aumenta el metabolismo hepático del cortisol, lo que contribuye a manifestar una falla adrenal latente.
Otro aspecto fundamental en estos síndromes es la sospecha y detección precoz de los componentes de cada uno de estos trastornos para evitar morbilidad e incluso mortalidad, por ejemplo evitar un debut de diabetes mellitus con cetoacidosis o hipotensión severa en el caso de una insuficiencia suprarrenal. Por tal motivo se recomienda realizar estudios funcionales con cierta frecuencia en pacientes que ya presentan uno o más de los componentes del síndrome. Por ejemplo, en un paciente portador de Diabetes Mellitus 1, siempre debe considerarse la posibilidad de un hipotiroidismo y/o una insuficiencia suprarrenal agregada, solicitando TSH, T4 libre, cortisol basal, electrolitos plasmáticos (ELP) y eventualmente un test de ACTH para cortisol.
Además de los estudios funcionales se puede adelantar aun más solicitando los autoanticuerpos involucrados, tal como se describen en la Tabla 4.
Los autoanticuerpos pueden estar presentes mucho antes de que se manifieste la enfermedad, en el caso de los anticuerpos anti 21 hidroxilasa están presentes en más del 90% de los pacientes con Enfermedad de Addison y su detección precede a la enfermedad en la mayoría (8). El diagnóstico de enfermedad de Addison en ausencia de estos anticuerpos requiere buscar otras causas como tuberculosis o adrenoleucodistrofia.
Aunque hoy es posible predecir la Diabetes tipo 1 con la detección de anticuerpos antiislotes de Langerhans y/o anti GAD (glutamato decarboxilasa), no es posible alterar su progresión. En cambio, detectando anticuerpos anti transglutaminasa en enfermedad celíaca si existe un cambio en la progresión de la enfermedad retirando el gluten de la dieta, lo que resulta relevante en el pronóstico del paciente.
En pacientes con diabetes mellitus tipo 1 que comienzan con episodios de hipoglicemia, especialmente nocturnas, o que sus requerimientos de insulina comienzan a bajar, hay que considerar la posibilidad de estar frente a un signo temprano de insuficiencia suprarrenal (7).
En el SPG 2, uno de cada siete parientes presentará un trastorno endocrino autoinmune, siendo la tiroiditis autoinmune la presentación más común, por lo que se recomienda estudio de función tiroidea y de anticuerpos antitiroideos con cierta frecuencia en esta población.
Finalmente, lo más importante para diagnosticar y tratar a tiempo a estos pacientes es sospechar precozmente que se está frente a un síndrome poliglandular autoinmune.
En cuanto al estudio genético, en la actualidad se recomienda sólo en SPG 1 con la detección del gen AIRE.
La autora declara no tener conflictos de interés, relacionados a este artículo.