





Las neoplasias endocrinas múltiples (NEM) tipo 1 y 2 son enfermedades genéticas heredadas en forma autosómica dominante. Las principales manifestaciones clínicas en NEM1 incluyen tumores paratiroideos, hipofisiarios y gastroenteropancreáticos. El test genético se puede realizar en los pacientes y potenciales portadores de mutaciones en el gen menin, pero la correlación genotipo-fenotipo es menos directa en comparación a NEM2. En la NEM2 el cáncer medular de tíroides (CMT) es común a los tres subtipos: NEM2A (feocromocitoma e hiperparatiroidismo), NEM2B (feocromocitoma y neuromas mucosos) y CMT familiar. A aquellos pacientes con mutación RET se les debe recomendar la realización de tiroidectomía profiláctica en la niñez, de acuerdo a la categoría de riesgo ATA. Algunos casos de CMT aparentemente esporádicos son actualmente NEM2 después de la realización del estudio genético para proto-oncogen RET, por lo tanto se recomienda la aplicación rutinaria de este estudio a todos los pacientes con CMT aparentemente esporádico.
Multiple endocrine neoplasia (MEN) type 1 and 2, are genetic diseases heritage in an autosomal trait. The major clinical manifestations in MEN1 include parathyroid, pituitary and gastroenteropancreatic neuroendocrine tumors. Genetic testing can be done in patients and potential carrier of the menin gene mutation, but the genotype-phenotype correlation is less straightforward in comparison with MEN2. In MEN2 the medullary thyroid carcinoma (MTC) is common to the three subtypes: MEN2A (phechromocytoma and hyperparathyroidism), MEN2B (phechromocytoma and mucosal neuromas) and familial MTC. Those with the RET mutation should be advised to have prophylactic thyroidectomy in the childhood, according with the ATA risk level. Some cases of apparently sporadic MTC are actually MEN2 after RET proto-oncogene testing is done, therefore routine application of this test is recommended in all cases of apparently sporadic MTC.
Las neoplasias endocrinas múltiples son síndromes clínicos de origen genético de herencia autosómica dominante que se manifiestan clínicamente por presencia de múltiples tumores benignos y malignos que involucran a órganos endocrinos y no endocrinos. Se han descrito 2 síndromes: neoplasia endocrina múltiple tipo 1(NEM1) y neoplasia endocrina múltiple tipo 2 (NEM2), ambos de baja prevalencia, pero cuya identificación es crucial, dado que presentan alta incidencia de tumores a edades tempranas con importante morbimortalidad. En relación a esto es muy importante el avance conseguido en las últimas décadas en relación a la identificación de los genes responsables de estas enfermedades, lo que ha permitido lograr un conocimiento más cabal de su fisiopatología y a la vez poder establecer grupos de riesgo, lo que es fundamental para el seguimiento y tratamiento de estos pacientes, principalmente en la NEM2.
El objetivo de esta revisión es describir estos síndromes desde un punto de vista fisiopatológico y clínico, con énfasis en la sospecha y en el seguimiento de los pacientes y sus familiares.
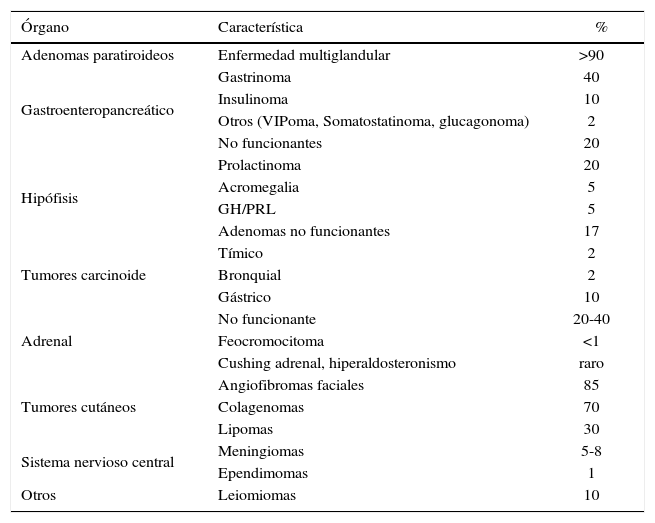
Neoplasia endocrina múltiple tipo 1La NEM1 (MIM # 131100) es una enfermedad autosómica dominante que predispone al desarrollo de tumores en la glándula paratiroides (Hiperparatiroidismo, HPP) (cercano al 100% a los 50 años), tumores éntero-pancreáticos (30-75%) y tumores de la adenohipófisis (10-60%) (1, 2). Algunos pacientes pueden desarrollar además tumores de la corteza adrenal, tumores carcinoides, angiofibromas faciales, colagenomas, lipomas, con una combinación variable de más de 20 tumores tumores endocrinos y no endocrinos (Tabla 1) (3, 4).
Tumores endocrinos y no endocrinos relacionados a nem1 (prevalencia a los 40 años)*
| Órgano | Característica | % |
|---|---|---|
| Adenomas paratiroideos | Enfermedad multiglandular | >90 |
| Gastroenteropancreático | Gastrinoma | 40 |
| Insulinoma | 10 | |
| Otros (VIPoma, Somatostatinoma, glucagonoma) | 2 | |
| No funcionantes | 20 | |
| Hipófisis | Prolactinoma | 20 |
| Acromegalia | 5 | |
| GH/PRL | 5 | |
| Adenomas no funcionantes | 17 | |
| Tumores carcinoide | Tímico | 2 |
| Bronquial | 2 | |
| Gástrico | 10 | |
| Adrenal | No funcionante | 20-40 |
| Feocromocitoma | <1 | |
| Cushing adrenal, hiperaldosteronismo | raro | |
| Tumores cutáneos | Angiofibromas faciales | 85 |
| Colagenomas | 70 | |
| Lipomas | 30 | |
| Sistema nervioso central | Meningiomas | 5-8 |
| Ependimomas | 1 | |
| Otros | Leiomiomas | 10 |
VIP, péptido intestinal vasoactivo ; GH, hormona de crecimiento; PRL, prolactina.
La NEM1 es una enfermedad rara y su prevalencia ha sido estimada en 2-3 casos 100.000 habs, causada por una mutación en el gen menin (MIM 613733) ubicado en el cromosoma 11q13 y es caracterizado por una alta penetrancia (94% en la 5ª década de la vida), alta mortalidad y una correlación genotipo-fenotipo no tan clara como ocurre en la NEM2. Se han descrito mutaciones de novo en hasta un 10% de los casos (5-10). El gen de la NEM1 consta de 10 exones que codifican una proteína de 610 aminoácidos llamada menin (menina, una proteína nuclear ampliamente expresada. La proteína menin es una proteína supresora de tumores y también juega un rol en la mantención de la estabilidad del DNA y regulación génica, sin embargo sus interacciones con otras proteínas así como su real mecanismo de supresión tumoral está aún en estudio. Se han descrito más de 1000 mutaciones de línea germinal, las cuales se encuentran ampliamente distribuidas a lo largo de todo el gen, involucrando las regiones codificadoras (exones) y sitios de splicing (unión del exón con el intrón) (10). En el modelo de tumorogénesis en NEM1 se aplica la “teoría de los dos golpes” de Knudson: una copia mutada de NEM1 es heredada a nivel de línea germinal del progenitor afectado (“primer golpe”), mientras que la copia normal (“wild-type”) es mutada a nivel somático (“segundo golpe”), generalmente con gran pérdida del material cromosómico (deleción), resultando en el desarrollo de un tumor. La presencia de este mecanismo es avalado por la presencia de pérdida de heterocigocidad (“loss of heterozygosity”, LOH) cuando el DNA tumoral es comparado con el DNA constitutivo (3).
El diagnóstico clínico de la NEM1 debe considerarse cuando se presentan 2 de los 3 principales tumores asociados a la NEM1 (tumores de las paratiroides, entero-pancreáticos e hipófisis). El diagnóstico de la NEM1 familiar se define como un caso índice NEM1 asociado a la presencia de un familiar de primer grado que tiene al menos uno de los tres tumores clásicos asociados a la NEM1. Otra forma de hacer el diagnóstico es cuando en un individuo sin manifestaciones clínicas se constata la presencia de mutación en el gen Menin (2, 5).
El estudio genético debe ofrecerse a los pacientes que cumplen los criterios clínicos previamente señalados (esporádico o familiar), sus familiares asintomáticos y pacientes que no cumplen criterios de NEM1, pero que tienen características sospechosas de NEM1, por ejemplo, hiperparatiroidismo recurrente, presencia de gastrinoma, tumor hipofisiario a temprana edad, tumores múltiples pancreáticos y tumores carcinoides. No se ha podido establecer una relación genotipo-fenotipo como ocurre en la NEM2, encontrándose presentaciones polimorfas en pacientes que presentan la misma mutación. Teniendo presente lo anteriormente expuesto, los principales beneficios de realizar estudio genético consisten en confirmar diagnóstico (descartando la presencia de fenocopias) y descartar la presencia de la enfermedad en aquellos familiares que no presentan la mutación, evitando de esta forma el seguimiento exhaustivo que estos pacientes requieren, disminuyendo la ansiedad y stress personal y familiar (11).
Los tumores paratiroideos son la primera manifestación de la NEM1 en más del 87% de los pacientes, sin embargo, otros tumores como los tumores pancreáticos o adenomas hipofisiarios pueden ser la primera manifestación (8). Los tumores paratiroideos presentan una alta penetrancia (cercana al 100% a los 50 años), correspondiendo al 2-4% de todos los hiperparatiroidismos (12). Habitualmente se presentan a edad más temprana que los casos esporádicos (20-25 v/s 60 años) y corresponde habitualmente a enfermedad multiglandular, lo cual sumado a la importante prevalencia de glándulas ectópicas le otorga al HPP asociado a NEM1 un alto riesgo de recurrencia (2). El tratamiento es quirúrgico y requiere exploración cervical bilateral por corresponder generalmente a enfermedad multiglandular, la extensión de la resección paratiroidea es discutible (paratiroidectomía subtotal v/s paratiroidectomía total + injerto autólogo, idealmente con medición de hormona paratiroidea intraoperatoria) y algunos centros realizan además timectomía de rutina para resección de eventuales glándulas paratiroides ectópicas y evitar el desarrollo de tumor carcinoide tímico, el cual es una causa de morbimortalidad importante en estos pacientes. El uso de calcimiméticos como el cinacalcet ha sido propuesto para el manejo médico de estos pacientes, pero aún la experiencia en pacientes con HPP asociado a NEM1 es escasa (13).
Los tumores gastroenteropancreáticos (TGEP) se presentan en el 30-80% de los pacientes con NEM1, pueden ser funcionales o no y típicamente se presentan a edades más tempranas que en los casos esporádicos, habitualmente son múltiples y son la primera causa de mortalidad en estos pacientes debido a su potencial comportamiento maligno. Dentro de los TGEP asociados a NEM1 destacan los gastrinomas, que representan alrededor del 50% de los TGEP asociados a NEM1; de todos los pacientes con gastrinomas alrededor de un 20% tendrá una NEM1. Son pequeños, rara vez se encuentran en el páncreas, siendo su localización habitual el duodeno y frecuentemente metastizan a hígado siendo muy importante su pesquisa precoz con marcadores serológicos (gastrina sérica). Es importante sospechar NEM1 en todo gastrinoma, especialmente cuando el diagnóstico es a edad temprana (2).
Otros tumores funcionantes son insulinomas, glucagonoma y VIPoma que presentan sindromes clínicos habitualmente de fácil reconocimiento. Existe una muy alta prevalencia de TGEP no funcionantes, principalmente pancreáticos, lo cual hace el estudio localizatorio de TGEP funcionantes bastante complejo, recurriendo a técnicas imagenológicas como tomografía axial computarizada (TAC), resonancia magnética (RM), PET/CT con análogos de somatostatina marcados con radioisótopos (por ejemplo 68 Ga), endosonografía, entre otros. Con respecto al manejo de los TGEP no funcionantes, poseen un importante potencial maligno por lo que la mayoría de los autores aconsejan resección con tamaño mayor a 2-3cm, pero este valor es tema de controversia en la literatura (14, 15).
El compromiso hipofisiario se presenta en 15-50% de los casos, se suele presentar a edades más precoces que los casos esporádicos, pero su comportamiento clínico no difiere mayormente de éstos, aunque algunos autores han reportado mayor agresividad. El tumor más frecuentemente encontrado es el prolactinoma (alrededor del 60% de los casos), siguiendo en frecuencia acromegalia. El tratamiento no difiere de los tumores hipofisiarios esporádicos y se debe sospechar la presencia de NEM1 cuando se presentan a edad temprana y cuando existe HPP asociado (16, 17).
Además de los tumores descritos existen otros tumores que también se presentan con mayor frecuencia en NEM1 como son los tumores carcinoides, principalmente de timo, bronquios, estómago y duodeno, los cuales representan la segunda causa de mortalidad en NEM1(2).
Las lesiones cutáneas en NEM1 son también frecuentes y deben ser consideradas como un elemento más de sospecha, los más frecuentes angiofibroma facial (64-88%), los colagenomas (63-72%) y lipomas (3-34%)(18).
El compromiso adrenal se presenta en 9-55% de los pacientes, siendo bilateral en el 12.5% de los casos, tanto funcionales como no funcionales, incluso hay casos descritos de carcinoma suprarrenal, por lo que este compromiso también debe ser buscado en forma dirigida (19).
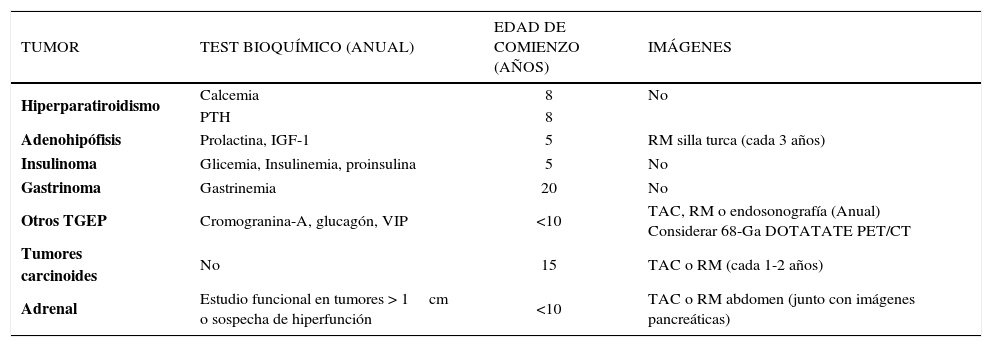
El seguimiento de estos pacientes es complejo y demandante ya que debido a la clínica variable y a la ausencia de relación genotipo-fenotipo, deber ser sometidos a múltiples exámenes de laboratorio e imagenológicos desde temprana edad para realizar pesquisa precoz (Tabla 2), por lo que se hace necesario contar con estudio genético para descartar la enfermedad y evitar estudios, gastos y stress innecesarios (2).
Seguimiento clínico para pacientes con diagnóstico de nem1*
| TUMOR | TEST BIOQUÍMICO (ANUAL) | EDAD DE COMIENZO (AÑOS) | IMÁGENES |
|---|---|---|---|
| Hiperparatiroidismo | Calcemia | 8 | No |
| PTH | 8 | ||
| Adenohipófisis | Prolactina, IGF-1 | 5 | RM silla turca (cada 3 años) |
| Insulinoma | Glicemia, Insulinemia, proinsulina | 5 | No |
| Gastrinoma | Gastrinemia | 20 | No |
| Otros TGEP | Cromogranina-A, glucagón, VIP | <10 | TAC, RM o endosonografía (Anual) Considerar 68-Ga DOTATATE PET/CT |
| Tumores carcinoides | No | 15 | TAC o RM (cada 1-2 años) |
| Adrenal | Estudio funcional en tumores > 1cm o sospecha de hiperfunción | <10 | TAC o RM abdomen (junto con imágenes pancreáticas) |
La neoplasia endocrina múltiple tipo 2 (NEM2) es un sindrome de herencia autosómica dominante y afecta aproximadamente a 1 en 30.000 individuos (5, 20-22). Los principales componentes de este sindrome son cáncer medular de tiroides (CMT), feocromocitoma (FEO) e HPP. El proto-oncogen RET (MIM 164761) ubicado en el cromosoma q11.2 codifica una proteína receptor de membrana del tipo tirosina quinasa (Figura 1). Mutaciones de este proto-oncogen son responsables de los 3 subtipos de CMT hereditario: NEM2A 80% (MIM 171400), NEM2B 5% (MIM 162300) y CMT familiar 15% (CMTF, MIM 155240). El CMT es común a los 3 subtipos y el riesgo de desarrollar CMT es cercano al 100%. Los pacientes con NEM 2A/2B tienen un riesgo de 30-50% de desarrollar FEO, especialmente aquellos que son portadores de mutaciones en los codones 634, A883F y M918T (20-23). Existen características que diferencian ambos subtipos de NEM2: por una parte sólo los pacientes con NEM2A tienen riesgo de desarrollar HPP y amiloidosis cutánea liquenificada (ACL) (10-30% y 12% respectivamente) y sólo los pacientes con NEM2B tienen hábito marfanoide, neuromas en la lengua (Figura 2), labios y subconjuntivales, además de ganglioneuromas difusos del tracto gastrointestinal (22). Los pacientes afectados de CMTF sufren exclusivamente de CMT; para probar que un miembro particular de una familia tiene CMTF es necesario demostrar la ausencia de FEO o HPP en dos o más generaciones de su familia, la comprobación de una mutación RET asociada sólo a CMTF y la existencia de al menos 4 miembros de una familia afectada con CMT sin FEO ni HPP (21, 23). Excepcionalmente hay familias en las cuales coexiste la enfermedad de Hirschsprung y NEM2 o CMTF, las cuales se asocian con mutaciones en los codones 609, 611, 618 y 620 (23, 24). El CMT en el contexto de NEM2B se desarrolla a edad más temprana y tiene un curso más agresivo comparado con los otros dos subtipos de CMT hereditario. Durante el primer año de vida, las características clásicas de NEM2B pueden estar ausentes y hay que prestar atención a algunos signos precoces como la incapacidad para llorar con lágrimas y la constipación (25). Existe una estrecha correlación genotipo-fenotipo en NEM2 y la agresividad del CMT es fuertemente influenciada por la mutación RET específica del paciente. Por ejemplo, se ha confirmado presencia de CMT en biopsias de tiroidectomías en niños tan pequeños como 15 meses en NEM2A y en niños de 9 meses en NEM2B (mutación codón M918T) y compromiso ganglionar a edades de 5 y 2.7 años respectivamente (20, 26-28). De esta forma, la identificación de una mutación específica de RET en un paciente de riesgo, permite orientar el tamizaje y optimizar el manejo clínico y quirúrgico. En el año 2009, después de una extensa revisión de la literatura, la American Thyroid Association (ATA) actualizó el consenso publicado el año 2001 y desarrolló un sistema de categorización de riesgo de desarrollar CMT y los otros tumores según el tipo de mutación con el objetivo de ofrecer recomendaciones para la edad de la tiroidectomía profiláctica, predecir el fenotipo y establecer quién debe ser tamizado para búsqueda de FEO e HPP como se muestra en la Tabla 3, adaptada de las guías de la ATA y otras referencias (20, 21, 29). Son 4 los grupos de riesgo desde el menos agresivo (A), intermedio (B), agresivo (C), donde se encuentra las mutaciones más frecuentes, codón 634, hasta el más agresivo (D), que corresponde a NEM2B. Es importante destacar que un CMT aparentemente esporádico puede ser en realidad un CMT hereditario. Estudios internacionales y nacionales mostraron que un 5-7% de pacientes sin historia familiar de CMT resultan ser hereditarios (29, 30). Esto demuestra la importancia estudio genético en todo paciente con CMT.


Recomendaciones para tiroidectomía profiláctica y tamizaje de tumores asociados a nem2.*
| CATEGORÍA DE RIESGO ATA | D | C | B | A |
|---|---|---|---|---|
| CODON RET | 918, 883 | 634 | 609, 611, 618, 620, 630 | 768, 790, 804, 891 |
| Edad de tiroidectomía profiláctica | Tan pronto como sea posible y dentro del 1er año de vida | < 5 años | Considerar cirugía antes de los 5 años de edad. Se puede postergar si criterios exigentes son cumplidos**. | Se puede postergar cirugía después de los 5 años de edad si criterios exigentes son cumplidos** |
| Edad de tamizaje para FEo | Iniciar a los 8 años de edad y luego anualmente | Iniciar a los 8 años de edad y luego anualmente | Iniciar a los 20 años de edad y luego anualmente | Iniciar a los 20 años de edad y luego anualmente |
| Edad de tamizaje para Hpp | No | Iniciar a los 8 años de edad y luego anualmente | Iniciar a los 20 años de edad y luego anualmente | Iniciar a los 20 años de edad y luego anualmente |
Las NEM son síndromes hereditarios autosómicos dominantes. La aplicación de las técnicas de biología molecular ha hecho posible descubrir los genes involucrados en estos síndromes. La identificación de aquellos individuos que están en riesgo de desarrollar la enfermedad requiere tamizajes bioquímcos e imaginológicos periódicos para pesquisar el probable desarrollo de los tumores endocrinos que forman parte del síndrome. La identificación de aquellos familiares que no presentan la mutación, permite excluirlos de mayor estudio, dando tranquilidad a la familia y disminuyendo costos, siendo éste, la principal aplicación del test genético en NEM1. En el caso de identificar los portadores de las mutaciones en el proto-oncogen RET, se debe realizar la tiroidectomía profiláctica y el seguimiento de acuerdo con la categoría de riesgo ATA.
Los autores declaran no tener conflictos de interés, relacionados a este artículo.