






Los trastornos de la glándula suprarrenal son variados debido a las características histológicas y funcionales que ésta presenta y debido a su baja frecuencia no se cuenta con una amplia información epidemiológica.
En este capítulo se revisará el cuadro clínico del síndrome de Cushing, los exámenes recomendados en las últimas guías de práctica clínica para su diagnóstico, el diagnóstico diferencial de causas las ACTH dependientes e independientes, métodos de imágenes recomendados y opciones terapéuticas
En la insuficiencia suprarrenal primaria o enfermedad de Addison revisaremos el cuadro clínico, etiología más frecuente y la utilidad y limitaciones de los exámenes diagnósticos y terapias disponibles.
Finalmente, se revisarán algunos aspectos de un problema que se ha hecho cada vez más frecuente debido a la mejor calidad técnica de los exámenes de imágenes, los incidentalomas suprarrenales.
Adrenal disorders are diverse due to its histological and functional features. Because of their low frequency do not have a large epidemiological information.
In this chapter we focus on the clinical features of Cushing’s syndrome, the tests recommended in the current clinical guidelines for diagnosis, differential diagnosis of ACTH dependent and independent causes, imaging methods and recommended therapeutic options.
In primary adrenal insufficiency or Addison’s disease we review clinical aspects, some etiologic aspects and, utility and limitations of diagnostic tests and therapies.
Finally, we review some aspects of a problem that has become increasingly common due to improved technical quality of imaging methods, the adrenal incidentalomas.
Los trastornos de la glándula suprarrenal son variados debido a las características histológicas y funcionales que ésta presenta. La corteza suprarrenal es la encargada de la función esteroidogénica. La zona más interna, que corresponde a la reticulosa es donde se sintetizan los andrógenos suprarrenales; en la zona fascicular, se sintetizan los glucocorticoides y, en la zona más externa que es la glomerulosa, se sintetizan los mineralocorticoides. Las zonas reticulosa y fascicular responden al estímulo de la corticotropina hipofisiaria (ACTH) en cambio, la zona glomerulosa prácticamente no expresa al receptor de ACTH y su estímulo principal es la angiotensina. La medula suprarrenal, formada por células cromafines, sintetiza catecolaminas y, al igual que el sistema simpático, es capaz de responder al estrés.
En cada una de estas zonas se expresan o manifiestan diferentes tipos de patologías que pueden determinar hiper o hipofunción, hiperplasia o tumores, llevando a cuadros clínicos específicos para cada una
La baja frecuencia con que se manifiestan estas patologías, ha dificultado el contar con datos epidemiológicos amplios. Se cuenta con la información de grandes centros de referencia extranjeros a los que se hará mención al desarrollar cada cuadro pero, a la fecha no contamos con registros nacionales.
Este capítulo revisará el hipercortisolismo y su cuadro clínico, el síndrome de Cushing (SC) y a la insuficiencia suprarrenal primaria o enfermedad de Addison. También nos referiremos a las implicancias clínicas del incidentaloma suprarrenal.
1.-Sindrome de cushing (SC)Corresponde al conjunto de manifestaciones clínicas y las complicaciones metabólicas derivadas del exceso de cortisol en los tejidos.
Se puede dividir en exógeno o iatrogénico, por el uso de corticoterapia en altas dosis por tiempo prolongado o, endógeno, debido al aumento de la secreción de cortisol por las glándulas suprarrenales.
Para el SC endógeno se ha estimado una incidencia en población europea de 2-3 casos por millón de habitantes, predominando en el sexo femenino (1).
Desde el punto de vista clínico los pacientes con síndrome de Cushing (SC) pueden presentar un cuadro bien característico, siendo algunas de las manifestaciones más específicas que otras (Tabla 1). Pacientes obesos pueden tener un fenotipo similar y son algunos hallazgos como: plétora facial, miopatía proximal o estrías rojo oscuro de más de 1cm de ancho, las que hacen más probable el diagnóstico.
Manifestaciones clinicas del sindrome de cushing
| INESPECIFICAS |
| • Obesidad abdominal. |
| • Hipertensión arterial. |
| • Trastornos neuropsiquiátricos: depresión o psicosis. |
| • Acné e hirsutismo, consecuencias de la activación de la unidad pilosebácea por el glucocorticoide. |
| • Alteraciones menstruales: oligomenorrea o amenorrea, por la supresión del eje gonadal por el hipercortisolismo. |
| MENOS INESPECIFICAS |
| • Acúmulo de grasa en dorso alto y en los huecos supraclaviculares. |
| • Hiperpigmentación de pliegues y superficies extensoras en los pacientes en que existe un marcado aumento de ACTH. |
| • Osteopenia u osteoporosis. |
| ESPECIFICAS |
| • Cara redonda y pletórica. |
| • Atrofia muscular y miopatía proximal. |
| • Piel fina y atrófica, equímosis espontáneas. |
| • Estrías violáceas, anchas (>1cm) en abdomen, tórax y extremidades. |
También se pueden encontrar alteraciones en exámenes del laboratorio general: aneosinofilia, linfopenia, elevación de la glicemia (intolerancia a glucosa o diabetes), dislipidemia (hipertrigliceridemia) e hipokalemia.
1.1.-Exámenes confirmatorios del diagnóstico de sindrome de cushingUna vez establecida la sospecha clínica de SC, se debe confirmar la existencia de un hipercortisolismo por medio de exámenes bioquímicos. Los exámenes recomendados en las últimas guías de práctica clínica de la Sociedad Americana de Endocrinología (Endocrine Society) son: cortisol libre en orina de 24 horas (CLU), test de supresión con 1mg de dexametasona a las 23 h (test de Nugent) y cortisol salival nocturno (2).
cortisol libre urinario: A diferencia de la determinación de cortisol en plasma o suero, que mide el cortisol unido a la cortisol binding globulin (CBG), proteína transportadora de cortisol, el CLU consiste en la determinación del cortisol libre de su proteína ligante. Algunas series han establecido hasta una sensibilidad de 100% y especificidad de 98%, pero hay que considerar que hasta un 15% de los SC puede tener al menos 1 determinación dentro del rango normal, por lo cual, es aconsejable en los casos con alta sospecha clínica, realizar al menos 2 mediciones en muestras de orina recolectadas en días distintos. Es categórico de SC el hallazgo de un CLU aumentado dos o más veces sobre el límite superior del rango normal, el cual dependerá de la técnica o kit utilizado en cada laboratorio.
Al interpretar el resultado del CLU se debe tener en cuenta el volumen urinario. Volúmenes sobre 4 litros, pueden dar valores falsamente elevados. Debido a que la mayoría del cortisol filtrado es metabolizado o reabsorbido, una ingestión de líquidos aumentada determina un mayor volumen urinario, lo que puede reducir la fracción de cortisol que es metabolizado. Lo inverso ocurre con volúmenes urinarios bajos o en la IRC. Siempre se debe medir creatinina en la misma muestra para asegurarse que la recolección de orina fue completa.
El test de Nugent consiste en la medición de cortisol plasmático a la mañana siguiente de una dosis de dexametasona de 1mg administrada a las 23 h. La dexametasona es un corticoide sintético de acción prolongada, que es metabolizada por la citocromo P450 3A4 (CYP3A4) y que no es detectada por los métodos de laboratorio que miden cortisol. Tradicionalmente el punto de corte entre la normalidad y el hipercortisolismo era 5µg/dl pero, algunos SC pueden suprimir bajo este nivel, determinando un porcentaje no despreciable de falsos negativos. En la reunión de expertos para un consenso en el diagnóstico de síndrome de Cushing del año 2003, se propuso utilizar como punto de corte 1.8µg/dl, para aumentar la sensibilidad del test (3).
Como se mencionó anteriormente, la técnica de determinación de cortisol en sangre, mide el unido a la CBG, por lo tanto, al interpretar el examen se debe descartar condiciones que la aumentan como por ejemplo: uso de estrógenos, anticonceptivos orales y embarazo, que pueden determinar un falso positivo.
Otra situación que puede llevar a un resultado erróneo en el test de Nugent, es el uso de drogas que aumentan el metabolismo de la dexametasona, por aumento de la actividad de la CYP3A4 (rifampicina, barbitúricos, carbamazepina, fenitoína, pioglitazona y algunos agonistas serotoninérgicos: citalopram y buspirona) (4).
cortisol salival nocturno (23 horas): en condiciones normales el cortisol alcanza su valor más alto alrededor de las 8 AM y su valor más bajo cercano a la medianoche (ritmo circadiano).
El cortisol matinal ha demostrado tener poca utilidad en el diagnóstico del SC ya que, aproximadamente un 60% de ellos pueden presentar un valor dentro del rango normal por lo tanto esta determinación no se recomienda (2, 3). En el hipercortisolismo endógeno de cualquier etiología, el cortisol de las 23h se encuentra elevado. El inconveniente para esta medición, es que, el paciente debería estar en una condición libre de estrés al momento de la toma de muestra. Para solucionar este problema, desde hace algún tiempo se introdujo el uso de la medición del cortisol en saliva, lo que permite realizar el examen sin que el paciente salga de su casa y además, tiene la ventaja que mide el cortisol libre de la proteína transportadora. Por otra parte, el cortisol permanece estable en la muestra de saliva hasta por una semana, lo que facilita su entrega al laboratorio (5, 6). Esta determinación ha sido propuesta como el examen de screening para el diagnóstico de SC. Un valor de cortisol >8.6nmol/L se considera como diagnóstico probable (3). Sin embargo, esta determinación puede tener variaciones dependiendo de la técnica utilizada y de la población estudiada por lo tanto, cada laboratorio debería establecer el rango normal (7). En nuestro país, esta determinación no está implementada de rutina en todos los centros.
El test de Liddle o test de supresión con dosis bajas de dexametasona, se puede utilizar cuando hay inconsistencias entre los resultados del cortisol libre urinario y el test de Nugent. Consiste en la administración de una dosis de dexametasona de 0.5mg cada 6 horas durante dos días.
Se debe realizar una recolección de orina de 24h previo al inicio de la dexametasona y otra al segundo día. Un valor menor de 36µg /24h se observa en sujetos normales. El inconveniente de esta prueba es que sólo alcanza alrededor de un 55% de sensibilidad para discriminar entre sujetos normales y con S. de Cushing. Utilizando la medición de cortisol en suero 6 horas después de la última dosis de dexametasona, la sensibilidad aumenta sobre 90% aplicando el punto de corte de 1.8µg/dl (8). Es poco recomendable su uso como test diagnóstico de primera línea (9).
Cuando dos pruebas se encuentran alteradas, el diagnóstico de SC es seguro y el paciente debe ser derivado al especialista para realizar el estudio etiológico. Cuando la clínica es sugerente pero, sólo una prueba está alterada, se habla de pseudocushing. Estos casos corresponden a sujetos que tienen una hiperactividad del eje hipotálamo-hipófisissuprarrenal pero, no un hipercortisolismo autónomo. Generalmente corresponden a pacientes con patología psiquiátrica como depresión o alcoholismo entre otras. Estos casos también deben ser derivados al especialista para confirmar o descartar el diagnóstico de SC.
1.2.-diagnóstico etiológicoEl SC endógeno se puede clasificar en ACTH dependiente y ACTH independiente (Tabla 2).
Causas de sindrome de cushing y frecuencia relativa
| ACTH dependiente |
| • Adenoma hipofisiario productor de ACTH o enfermedad de Cushing (75%). |
| • Tumores productores de ACTH: pulmón, timo, páncreas, carcinoma medular de tiroides, feocromocitoma (~ 10%) |
| ACTH independiente: |
| • Adenoma suprarrenal (~ 15%) |
| • Carcinoma suprarrenal (<1%) |
| • Hiperplasia suprarrenal bilateral micro o macronodular (<1%) |
Aunque la causa más frecuente es la hipofisiaria o enfermedad de Cushing (EC), que corresponde aproximadamente a un 75% de los casos, siempre se debe realizar el estudio que se detalla a continuación antes de solicitar los estudios de imágenes.
La determinación de ACTH plasmática en una muestra matinal permite orientarse a una causa ACTH independiente o suprarrenal, en que se encontrarán valores normales bajos o suprimidos o, ACTH dependiente, en los cuales se encontrará aumentada o, inapropiadamente normal para el nivel de hipercortisolismo.
Cuando se sospecha una causa ACTH dependiente se debería aplicar test funcionales que permitan discriminar entre una EC y un tumor productor de ACTH ectópico.
Los test que han mostrado utilidad en el diagnóstico diferencial son: la supresión con dosis altas de dexametasona y la prueba de estimulación con un secretagogo de ACTH.
En el test de tyrrel o, supresión con 8mg de dexametasona administrada a las 23h, los casos con EC presentan una supresión del cortisol plasmático de más del 50% respecto del valor basal, porque los corticotropos tumorales conservan la capacidad de suprimir con dosis altas de corticoides. Sin embargo, se ha observado que algunos casos de ACTH ectópico pueden suprimir por lo que recomendamos que no sea utilizada como único test diagnóstico (10).
Para el test de estimulación se recomienda el uso de factor liberador de corticotropina (CRH). En EC se observa un aumento de la ACTH y del cortisol como respuesta al estímulo, lo que no se observa en el caso del tumor ectópico productor de ACTH que, habitualmente no expresa el receptor para CRH.
Algunos autores han utilizado como alternativa de estímulo la vasopresina, o su análogo desmopresina (DDAVP), por su mayor disponibilidad y menor costo que el CRH. El receptor V3 también es expresado por los corticotropos determinando respuesta de ACTH frente a este estímulo. Sin embargo, aún no hay consenso para su utilización en el diagnóstico diferencial del SC ACTH dependiente (2).
La cateterización de senos petrosos con medición de ACTH central y periférica, está indicado para diferenciar entre ACTH ectópico y de causa hipofisiaria, cuando las pruebas funcionales o las imágenes no son concluyentes por lo que se reserva para casos especiales.
En la última década, algunos autores han propuesto no utilizar las pruebas funcionales, porque han encontrado un porcentaje no despreciable de falsos positivos y negativos y recomiendan directamente realizar una resonancia nuclear magnética (RNM) de silla turca y, si esta es negativa o dudosa, realizar un cateterismo de senos petrosos (11, 12). Sin embargo, no lo recomendamos porque hay que considerar que hasta en un 10% de las RNM pueden encontrarse incidentalomas hipofisiarios y que, para el cateterismo de senos petrosos se requiere de infraestructura, personal especializado y además, tiene un elevado costo, sobre todo en nuestro medio. Por lo tanto, es de resorte del especialista solicitar e interpretar las pruebas funcionales que orienten a la causa del SC ACTH dependiente, para luego solicitar los exámenes de imágenes precisos y evitar errores terapéuticos.
1.3.-Métodos RadiológicosEn el caso que los test funcionales confirmen la sospecha de una EC, el examen indicado es la RNM de silla turca con contraste, que permite detectar adenomas pequeños.
En el caso que la sospecha de la etiología sea una localización suprarrenal, los exámenes utilizados son la tomografía computada de abdomen o la RNM de suprarrenales. La TAC de abdomen con contraste y cortes finos a nivel de la suprarrenal continúa siendo un examen sensible y de menor costo.
Cuando se sospecha un tumor productor de ACTH ectópico, se debe indicar en primer lugar una tomografía computada de tórax por ser ésta la ubicación más frecuente de estos tumores (carcinomas de células pequeñas, carcinoides bronquiales o de timo). En los casos en que no se logra localizar el tumor o hay sospecha de diseminación, está indicado realizar una tomografía por emisión de positrones (PET).
1.4.-tratamientoEn el SC de cualquier etiología el tratamiento inicial es quirúrgico
La mayoría de los casos de EC corresponden a un microadenoma y la indicación es una resección transesfenoidal del tumor (RTE). El éxito quirúrgico varía en las distintas series entre 50-90%, dependiendo del tamaño del adenoma y de la experiencia del equipo quirúrgico En los casos no curados otras alternativas terapéuticas son la radioterapia o la suprarrenalectomía bilateral (12).
Recientemente se aprobó el uso de Pasireotide, un análogo de somatostatina, para el tratamiento de la EC cuando no hay curación después de la cirugía o, ésta está contraindicada (13).
Los tumores suprarrenales son mayoritariamente benignos y en estos casos está indicada una suprarrenalectomía unilateral, a diferencia de la hiperplasia nodular en que se debe realizar una suprarrenalectomia bilateral.
En el carcinoma suprarrenal con extensión regional o a distancia, se pueden utilizar terapias antiglucocorticoides (aminoglutetimida, ketoconazol, Mifepristone (RU486), mitotane). La aminoglutetimida y el ketoconazol actúan bloqueando enzimas citocromo p450 de la vía de síntesis del cortisol. Después de un tiempo de uso se puede observar un aumento de los niveles de cortisol debido a la disminución de la supresión de la ACTH, que comienza a estimular nuevamente a la suprarrenal. El RU-486 es una droga anti-progestacional que, a altas dosis, compite con los glucocorticoides por la unión a su receptor, bloqueando su acción. También se ha aprobado su uso en EC fundamentalmente cuando se asocia a diabetes de difícil manejo. (14)
El mitotane es un compuesto órgano clorado derivado del DDT, con efecto adrenolítico. Actúa a nivel mitocondrial, llevando a la destrucción de las mitocondrias y necrosis de las células de la corteza produciendo una suprarrenalectomía médica. Actualmente, es la terapia coadyuvante recomendada para el carcinoma suprarrenal (15).
En los tumores ectópicos que pueden ser identificados, que son resecables y que no tienen metástasis, su extirpación suele ser curativa pero, en los casos en que no es localizable o, existe diseminación, estaría indicada la suprarrenalectomía bilateral, para evitar la progresión de las complicaciones derivadas del exceso de cortisol. Las terapias antiglucocorticoideas también pueden ser utilizadas en estos casos. El uso de análogos de somatostatina (Sandostatina), está indicada cuando se observa captación en el octreoscan, lo que indica presencia de receptores (12).
1.5.-PronósticoEl pronóstico del SC depende de un diagnóstico precoz y correcto y de la aplicación de una terapia adecuada. El hipercortisolismo crónico determina un deterioro en la calidad de vida y elevada morbi-mortalidad derivada de complicaciones metabólicas, eventos cardiovasculares, infecciones, fracturas patológicas y trastornos psiquiátricos de difícil tratamiento (14).
2.-Insuficiencia suprarrenal primaria o enfermedad de addisonEn la insuficiencia suprarrenal primaria, se produce una falla en la producción de todas las hormonas esteroidales, a diferencia de la insuficiencia suprarrenal secundaria en que sólo ocurre déficit de cortisol por la falta del estímulo de ACTH. Es más frecuente en mujeres, en la cuarta década de la vida. La prevalencia es 100 casos/millón y la incidencia, 5,5 casos/millón, a diferencia de la secundaria que es más frecuente en mujeres, en la sexta década de la vida y con una prevalencia de 250 casos/millón (16).
El cuadro clínico se caracteriza por signos y síntomas inespecíficos como baja de peso, dolor abdominal, nausea, vómitos, diarrea, síntomas de hipoglicemia e hipotensión ortostática que, habitualmente se asocian a otros bien característicos como hiperpigmentación de piel, pliegues y mucosas, melanoplaquias, astenia y avidez por la sal. En la mujer, el déficit de andrógenos puede determinar disminución de fuerza, disminución del vello pubiano y axilar y, disminución de líbido.
Las causas de insuficiencia suprarrenal primaria se presentan en la Tabla 3. La etiología más frecuente en la actualidad es la autoinmune (90%). En diversos estudios se ha descrito que más de la mitad de los pacientes que presentan insuficiencia suprarrenal autoinmune pueden llegar a desarrollar un síndrome poliglandular autoinmune tipo 2, en el cual se asocian enfermedad de Addison, tiroiditis crónica y en alrededor del 50% de los casos, diabetes tipo 1. Se ha descrito que la enfermedad de Addison puede preceder hasta en 20 años a la aparición del compromiso poliglandular. Por otra parte, en pacientes con diabetes tipo 1, tiroiditis autoinmune o enfermedad de Graves, se debería buscar dirigidamente los síntomas y signos de insuficiencia suprarrenal y también, frente a la presencia de manifestaciones extra endocrinas de autoinmunidad como vitiligo, enfermedad celiaca o anemia perniciosa (17).
Causas de insuficiencia suprarrenal primaria o enfermedad de addison
| Autoinmune: |
| • Aislada, S. poliglandular autoinmune |
| Infecciones: |
| • Tuberculosis, Infección sistémica por hongos, SIDA |
| Infiltración tumoral: |
| • Linfoma, metástasis cáncer pulmón, mama, estómago, colon. |
| Infiltrativas: |
| • Amiloidosis, hemocromatosis, sarcoidosis |
| Infarto o hemorragia suprarrenal bilateral: |
| • Meningococcemia, sepsis, trauma, anticoagulante lúpico |
| Quirúrgica: |
| • Suprarrenalectomía total bilateral |
| Hereditarias (muy poco frecuentes): |
| • Adrenoleucodistrofia |
| • Hipoplasia congénita ligada a X |
En los exámenes generales hay algunos hallazgos que pueden orientar al diagnóstico, sobre todo si se asocian a la clínica: eosinofilia, hiponatremia, hiperkalemia, hipercalcemia sin hiperfosfemia, hipoglicemia.
La medición de cortisol basal suele no ser un parámetro útil, salvo cuando se encuentra bajo 3 µg/dl o, bajo 15µg/dl en una situación de estrés importante, estos valores son altamente sugerentes de falla suprarrenal. La gran mayoría de los casos en etapas iniciales pueden presentar niveles dentro del rango normal o normal bajo, lo que puede llevar a omitir el diagnóstico. Como ya se mencionó para el estudio del SC, al interpretar el cortisol en sangre, se debe considerar los factores que aumentan la CBG. Por esto, la medición de ACTH plasmática, también se debe realizar frente a la sospecha de insuficiencia suprarrenal ya que, en la insuficiencia primaria establecida estará por sobre el rango normal. También permite establecer si se trata de una insuficiencia primaria o secundaria (18, 19).
Aunque el patrón de oro para el diagnóstico de insuficiencia suprarrenal es la falta de elevación del cortisol después de inducir una hipoglicemia con insulina, por los riesgos asociados, esta prueba prácticamente no se utiliza y se acepta a la prueba de estimulación con 250µg de ACTH de acción rápida (cosintropina, Synacthen®), como método de confirmación diagnóstica. Esta prueba se debería aplicar cada vez que los valores de cortisol matinal descienden bajo la media del rango normal (18). La respuesta máxima a los 30 minutos del estímulo, varía en sujetos normales entre 18.5 y 23µg/dl, existiendo diferencias según el sexo y el método utilizado para medir el cortisol (20). El punto de corte de cortisol más utilizado en la actualidad es 18µg/dl a los 30 minutos post estímulo (19).
En nuestro medio, en algunos centros se utiliza la estimulación con ACTH de depósito. La prueba consiste en tomar un cortisol basal, inyectar intramuscularmente cosintropina depot y tomar otra muestra de cortisol 6 horas después. Los sujetos normales alcanzan en esta muestra un cortisol sobre 25µg/dl. Si no se alcanza este nivel, el paciente debe recibir 2 inyecciones más, en días consecutivos y tomar nueva muestra para determinación de cortisol 6 horas después de la última inyección. Si el cortisol está sobre 25µg/dl, se trata de una insuficiencia suprarrenal secundaria. Si no alcanza este valor, se confirma la insuficiencia suprarrenal primaria.
La medición de actividad de renina plasmática (ARP) tomada en posición supina también es útil en el diagnóstico de insuficiencia suprarrenal inicial. En estos casos, se encuentra aumentada debido a la disminución de los niveles de aldosterona. Las alteraciones electrolíticas, hiperkalemia e hiponatremia, generalmente son tardías y en etapas tempranas de la enfermedad pueden estar normales.
En cuanto al estudio etiológico, la medición de anticuerpos anti 21 hidroxilasa permite confirmar la etiología autoinmune. Cuando son negativos, se recomienda realizar algún examen de imágenes en busca de posibles focos infecciosos (TBC), procesos infiltrativos o hemorragia.
2.2.-TratamientoEn insuficiencia suprarrenal primaria se deben sustituir cortisol y mineralocorticoides. La sustitución de andrógenos como la dehidroepiandrosterona (DHEA), se ha preconizado principalmente para mejorar la calidad de vida, sobre todo en mujeres, aumentando fuerza muscular, libido y densidad mineral ósea (DMO) pero, hasta ahora, los resultados no han sido concluyentes y no se recomienda ampliamente su uso por la falta de un preparado farmacéutico con calidad certificada (21-23).
El corticoide a utilizar es la hidrocortisona, glucocorticoide de acción corta (vida media menor de 12h), que es el sustrato natural de la 11β Hidroxiesteroide dehidrogenasa (11βHSD) tipo 2. Esta enzima es la encargada de impedir la activación del receptor de mineralocorticoides por cortisol, inactivándolo a través de la conversión a cortisona pero, tiene baja capacidad inactivante sobre otros glucocorticoides.
La producción diaria de cortisol es 9.9mg/día, cantidad menor a la estimada previamente de 15mg. Esto se debe a que la 11βHSD tipo 1, convierte a la cortisona en cortisol, amplificando la señal en los tejidos blanco. Por lo tanto, el cálculo de la dosis debe hacerse por m2 de superficie corporal. La dosis de sustitución habitual va de 15-30mg/día. También hay que considerar que la secreción de cortisol tiene un ritmo pulsátil o ultradiano y uno circadiano por lo tanto, se debe simular este ritmo circadiano fraccionando la hidrocortisona en 2 y hasta 3 dosis. Se debe evitar la dosis nocturna para mantener un periodo de baja ocupación de los receptores en la noche. Recientemente se ha desarrollado una preparación de hidrocortisona de liberación modificada (Chronocort®), que permitiría realizar la sustitución en forma más fisiológica, simulando el aumento de la liberación del cortisol después de la medianoche pero, hasta ahora sólo se ha utilizado en ensayos clínicos (24).
Como no existe un buen examen para titular la dosis de hidrocortisona, la evaluación de la terapia es básicamente clínica, basada principalmente en el bienestar general del paciente, recuperación del peso, desaparición de la hiperpigmentación.
La sustitución mineralocorticoidea se realiza con fludrocortisona. La dosis diaria puede fluctuar entre 0,025 a 0,1mg. La titulación de la dosis se hace midiendo los niveles de sodio, potasio y ARP, la que nunca debe estar suprimida. Desde el punto de vista clínico, se debe vigilar la aparición de hipertensión arterial.
2.3.-Complicaciones de la sustitución glucocorticoidea densidad mineral ósea (DMO)Desde hace bastantes años, algunos autores han descrito en series pequeñas, que los pacientes con insuficiencia suprarrenal que utilizaban una dosis de sustitución de hidrocortisona de 30mg/día o mayor, presentaban una disminución de la DMO comparados con controles, principalmente en las mujeres postmenopáusicas. Recientemente, el análisis de 2 cohortes con más de 100 pacientes con enfermedad de Addison, que utilizaban una dosis promedio de hidrocortisona de 26,5mg/día demostró: una disminución significativa de la DMO en columna lumbar y cadera en los casos respecto de controles de la población; que hubo una correlación inversa de la DMO con la dosis del glucocorticoide y; que los pacientes que utilizaban un glucocorticoide sintético, tenían DMO más baja (25). Otro estudio también demostró que los pacientes que utilizan dosis más bajas de glucocorticoide presentaban una DMO normal y que el uso de un glucocorticoide de acción prolongada determina una disminución significativa de la DMO (26).
Metabolismo de la glucosaFisiológicamente los glucocorticoides pueden aumentar la glicemia e insulinemia postprandiales. En pacientes con enfermedad de Addison, son pocos los estudios que han evaluado en un número importante de casos el efecto sobre el metabolismo de la glucosa pero, se ha descrito mayor porcentaje de obesidad abdominal, intolerancia a la glucosa y dislipidemia en estos casos comparados con controles de la misma edad (27).
Como vigilancia de la terapia de sustitución, se debe evaluar la aparición de complicaciones metabólicas además, de la repercusión sobre la DMO.
2.4.-MortalidadDesde el advenimiento de la terapia de sustitución con glucocorticoides para la insuficiencia suprarrenal, se consideró que la expectativa de vida de estos pacientes era igual a la población general. Sin embargo, en pacientes con insuficiencia secundaria a hipopituitarismo, la mortalidad observada es mayor que la esperada pero, asociada principalmente al déficit de gonadotrofinas (28).
Un estudio poblacional realizado en Suecia, demostró en más de 1000 pacientes con enfermedad de Addison, que el riesgo de muerte era más de 2 veces mayor que en la población general, tanto para hombres como para mujeres, siendo las causas cardiovasculares, infecciosas y neoplasias malignas, las principales causas de muerte (29). Posteriormente, un estudio realizado en Noruega, no encontró diferencias en la tasa de mortalidad excepto para los casos diagnosticados antes de los 40 años, siendo la insuficiencia suprarrenal aguda y las infecciones, las principales causas, especialmente en hombres (30).
La mortalidad por una crisis suprarrenal en un paciente ya diagnosticado es perfectamente prevenible. En primer lugar, es fundamental instruir al paciente en no disminuir la dosis de hidrocortisona sin la indicación de un especialista y sobre la conducta a seguir frente a situaciones de estrés. El paciente y sus familiares deben ser instruidos en el ajuste de dosis frente a estas situaciones. Generalmente, la dosis debe aumentarse al doble durante enfermedades intercurrentes como infecciones respiratorias con fiebre. En el caso de infecciones gastrointestinales severas, se debería administrar una dosis parenteral. En el caso de cirugías mayores, traumas, trabajo de parto o patologías que requieran hospitalización en unidades de cuidados intermedios o intensivos, debe administrarse dosis de estrés de 50-100mg ev. c/8 h hasta superar la emergencia. El paciente siempre debería portar un brazalete o tarjeta de advertencia con su diagnóstico y, en caso de viajes o permanencia en lugares alejados de asistencia médica, debería tener un kit para inyección IM de 100mg de hidrocortisona.
3.-IncidentalomaSe define como “incidentaloma” suprarrenal a una masa mayor o igual a 1cm de diámetro que se descubre incidentalmente en un examen de imágenes abdominal o torácico, no realizado para etapificación o seguimiento de un cáncer; en un paciente libre de síntomas o signos sugerentes de enfermedad suprarrenal (31).
Debido a la mejor calidad técnica de los exámenes de imágenes y el fácil acceso a ellos, el hallazgo de incidentalomas suprarrenales se ha hecho cada vez más frecuente. La probabilidad de encontrar un incidentaloma suprarrenal aumenta con la edad, alcanzando una frecuencia de hasta 10% entre los 50 y 70 años, en cambio en niños no supera el 0,4% (32).
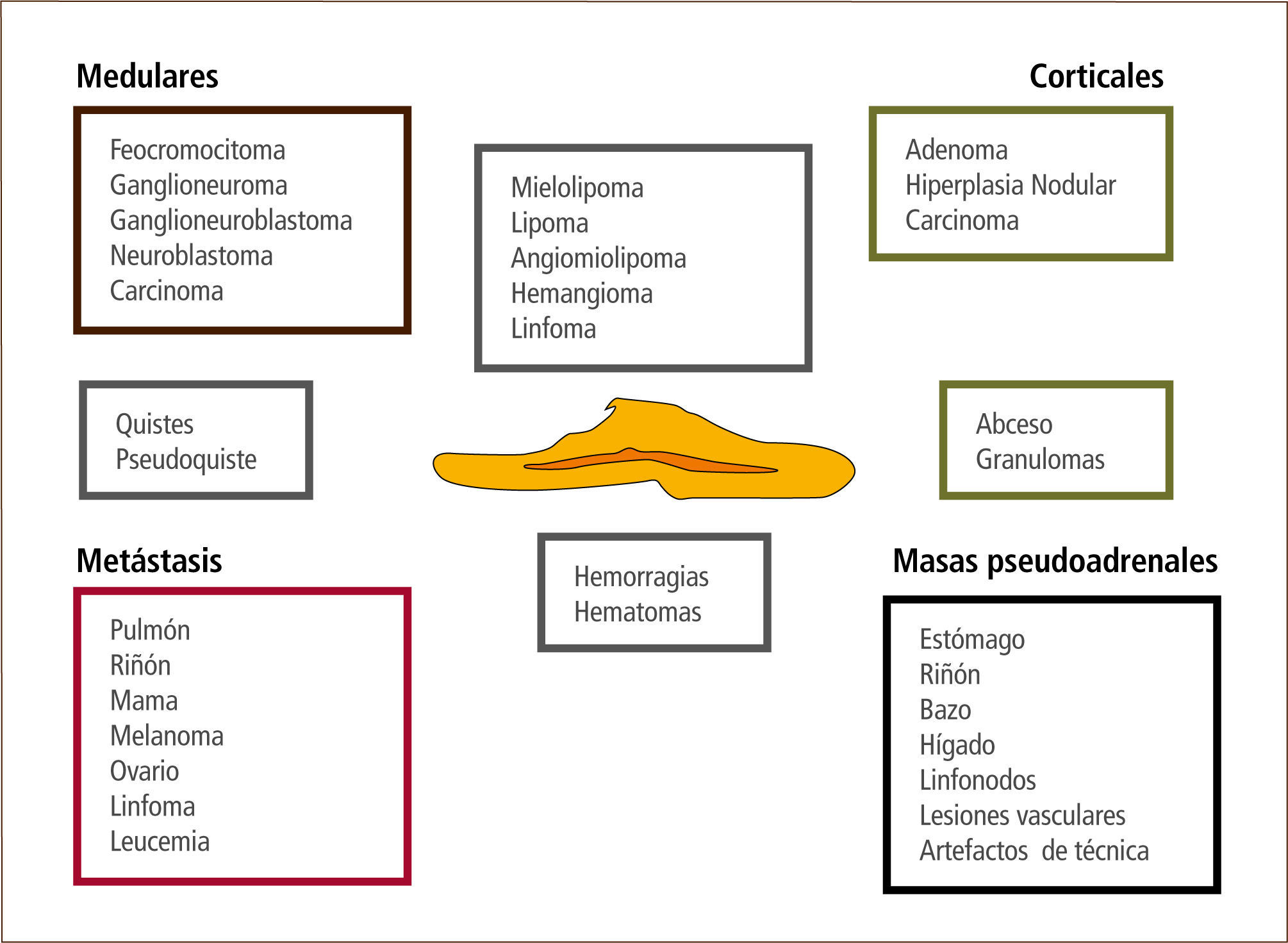
La gran mayoría corresponde a nódulos menores de 4cm “no funcionantes” o que no producen hormonas. Sólo un 20% suele ser funcionante al momento del diagnóstico y alrededor de 80% corresponde a tumores benignos o adenomas (32). La figura 1 muestra los distintos tipos de lesiones que pueden presentarse como incidentalomas suprarrenales.
3.1.-Estudio del incidentaloma suprarrenal
Es fundamental determinar la naturaleza de la masa para definir si el paciente requerirá un tratamiento quirúrgico o no.
Evaluación por imágenesLa tomografía computada de abdomen de alta resolución es un examen que puede aportar información útil en el diagnóstico diferencial, evaluando las unidades Hounsfield (UH) precontraste y el lavado del medio de contraste. Los adenomas presentan una forma regular, márgenes definidos, contenido homogéneo e hipodenso (< 10 UH), con fase de lavado del contraste rápida. En cambio los carcinomas presentan una forma irregular, márgenes mal definidos, necrosis y hemorragia intratumoral, aumento de la vascularización, contenido inhomogéneo e hiperdenso (> 10 UH) y fase de lavado del contraste retardado. Por otra parte, los feocromocitomas presentan márgenes bien definidos, aumento de la vascularización, zonas quísticas y hemorragia intratumoral, contenido inhomogéneo e hiperdenso y fase de lavado del contraste retardado. Las metástasis de otros órganos presentan una forma irregular, márgenes mal definidos, contenido inhomogéneo e hiperdenso (> 10 UH) y fase de lavado del contraste retardada (33).
Evaluación hormonalSi bien no hay un consenso sobre la aproximación diagnóstica óptima, los expertos recomiendan realizar en todos los pacientes una evaluación hormonal para determinar la funcionalidad del nódulo excepto, cuando la imagen radiológica es concluyente de mielolipoma o quiste (31).
En todos los casos se debe descartar feocromocitoma realizando una determinación de catecolaminas en orina de 24 horas o en plasma, cuando la recolección de orina es difícil o, hay diuresis disminuida. En todo paciente hipertenso o con hipokalemia se debe medir además aldosterona y actividad de renina plasmática. En cuanto a la evaluación del hipercortisolismo algunos expertos proponen realizarla sólo en los casos en que hay hechos consistentes con un SC como aumento de peso, intolerancia a la glucosa u osteopenia inexplicada (34). Sin embargo otros grupos proponen evaluarlo en todos los pacientes (35). Los exámenes indicados son el test de Nugent y la medición de cortisol salival nocturno. Hasta ahora no existe un consenso respecto del punto de corte en la supresión con dexametasona en estos casos. Un valor menor de 1.8µg/ml descarta un hipercortisolismo y un valor mayor de 5µg/ml lo confirma. Valores intermedios son considerados inciertos pero, en nuestra opinión, cualquier resultado superior a 1.8µg/ml obliga a completar el estudio y realizar un seguimiento estrecho del paciente. En general, estos casos son considerados como SC subclínicos.
Punción con aguja finaLa citología no es útil para discriminar entre un adenoma y un carcinoma suprarrenal, debido a que aún no hay un marcador citológico de malignidad. Además, tiene complicaciones como hemotórax o hematoma hepático y, aunque infrecuente, la punción de una lesión maligna puede resultar en siembra de células tumorales y la reacción inflamatoria puede dificultar la eventual cirugía (36). En la gran mayoría de los casos de incidentaloma la punción es no diagnóstica y sólo estaría indicada frente a un paciente con cáncer o sospecha de tumor de otro origen o, cuando hay sospecha de una infección o absceso (37).
3.2.-Indicación quirúrgicaEn todos los casos en que la evaluación hormonal demuestra feocromocitoma o hiperaldosteronismo por adenoma, está indicada la suprarrenalectomía. En los casos de hipercortisolismo o SC subclínico, las evidencias no son contundentes en cuanto a la indicación o no indicación quirúrgica ya que, no está bien establecida la capacidad de la cirugía de revertir los efectos de un hipercortisolismo leve (38, 39).
En los tumores no funcionantes es el tamaño el que determina la indicación. De acuerdo a los resultados de diferentes series publicadas, se establece que un tamaño de 4cm tiene la mayor sensibilidad para discriminar entre adenoma y carcinoma (40, 41). Sin embargo, hay que considerar que, independiente del tamaño, no tiene indicación quirúrgica si la TAC es diagnóstica de mielolipoma o quiste y el paciente es asintomático o, se trata de un hematoma agudo o lesión tuberculosa. También, en sujetos muy añosos con tumores no funcionantes la opción quirúrgica no es recomendada.
3.3-SeguimientoDe acuerdo a las diferentes series, hasta 20% de los incidentalomas pueden aumentar de tamaño y hasta 25% puede llegar a transformarse en funcionante en un seguimiento a 5 años (32, 42).
Algunos autores recomiendan una evaluación anual de hiperfunción y control de TAC de suprarrenales a los 6, 12 y 24 meses (31, 34, 35). Otros autores, realizaron un metanálisis, evaluando los resultados a dos años de la aplicación de las recomendaciones vigentes, encontrando un riesgo bajo de detección de cáncer (1:1000) o desarrollo de SC subclínico (1:250) y que, el riesgo de un cáncer fatal inducido por la exposición a la radiación durante el seguimiento con TAC va de 1:430 a 1:2170. Por esta razón y por el elevado costo que éste implica, proponen no realizar seguimiento en las lesiones menores de 4cm y con aspecto radiológico benigno (43).
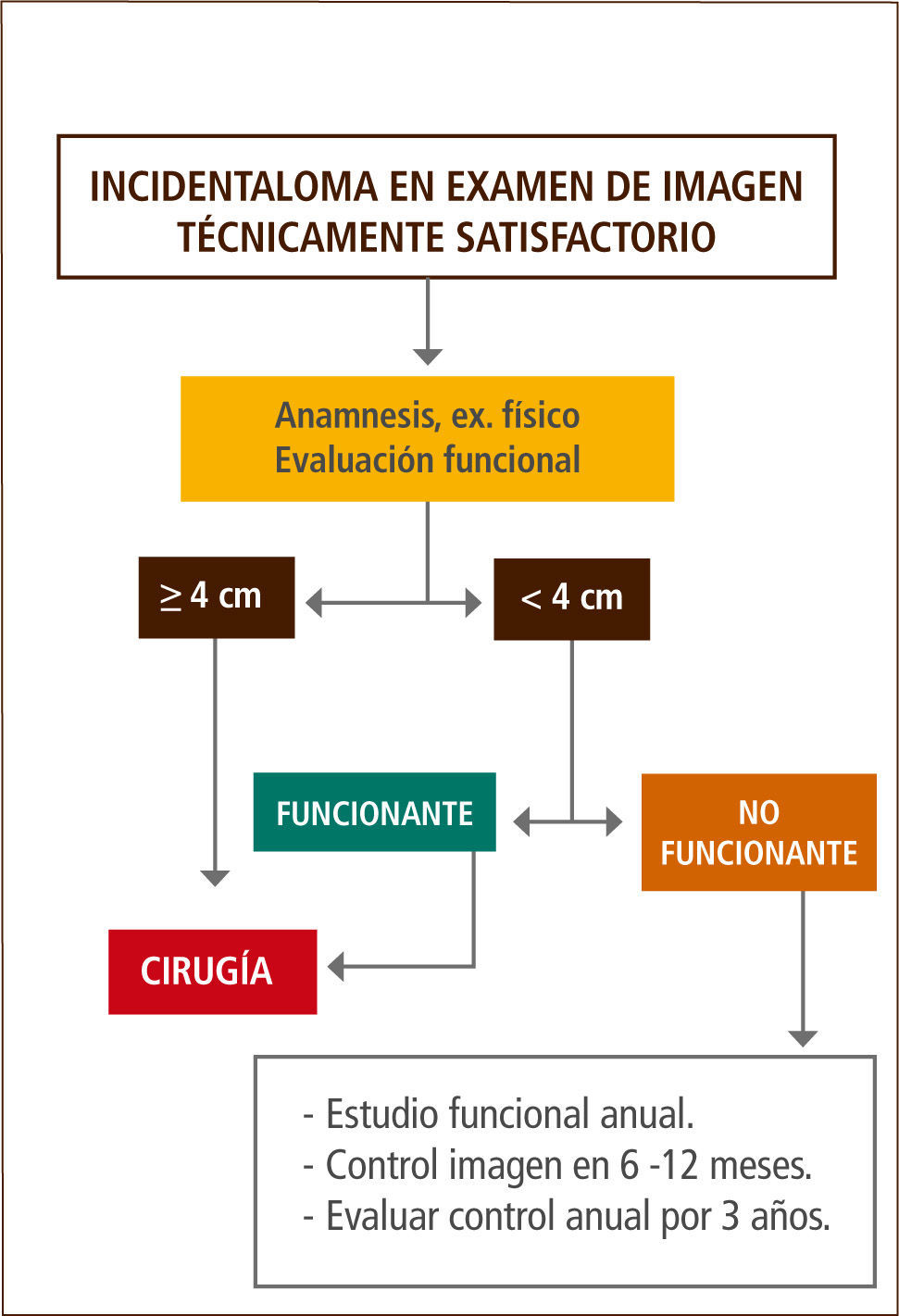
En nuestra opinión, los tumores menores de 4cm que presentan secreción de cortisol al momento del diagnóstico o del seguimiento, deben ir a cirugía exceptuando los casos que presenten alguna contraindicación debido a que, en nuestro medio es difícil mantener un seguimiento prolongado de los pacientes. En los tumores no funcionantes sugerimos un control anual bioquímico y de imágenes durante al menos 3 años (Figura 2).
Sintesis
La patología suprarrenal en general, aún constituye un desafío desde el punto de vista diagnóstico y terapéutico. Como consecuencia de su baja frecuencia, se hace difícil el estudio y análisis contemporáneo de un número significativo de casos. Probablemente los nuevos avances tecnológicos permitirán optimizar el enfrentamiento de estos pacientes.
La autora declara no tener conflictos de interés, relacionados a este artículo.