







La hipertensión arterial pulmonar (HAP) es una enfermedad crónica, que se caracteriza por el aumento de la resistencia vascular pulmonar (RVP) a nivel de la arteriola pulmonar, que provoca una progresiva sobrecarga y posterior disfunción del ventrículo derecho (VD), que en etapas finales lleva a la insuficiencia cardiaca derecha, la cual sella su pronóstico. La HAP es más frecuente en mujeres jóvenes en plena edad productiva, siendo la supervivencia media de 2-3 años, antes de la aparición de terapias específicas. La base genética sugiere una herencia autosómica dominante con penetrancia incompleta, reconociéndose principalmente la afección del BMPR2. En la etiopatogenia se reconoce una alteración en las señales que controlan fundamentalmente el equilibrio vasocontrictor-vasodilatador a nivel del endotelio, con un desbalance hacia la proliferación y vasoconstricción, en las que están involucradas 3 vías patogénicas: La del Óxido nítrico (ON), de la Prostaciclina (PG) y de la Endotelina (ET). El diagnóstico precoz de la HAP se asocia con una mejor supervivencia a largo plazo, por lo que su búsqueda ante un paciente con disnea, fatiga, dolor torácico y/o síncopes, así como en las poblaciones en riesgo, como son familiares en 1° con HAP, Esclerodermia y portadores de Hipertensión Portal, debería ser la estrategia de elección. La Ecocardiografía Doppler (ECO) es la herramienta de pesquisa más utilizada en la práctica clínica actual. El diagnóstico debe ser confirmado mediante un cateterismo derecho, con mediciones directas de la presión arterial pulmonar, y debe realizarse prueba de vasoreactividad. El advenimiento de los tratamientos farmacológicos-HAP específicos ha provocado un cambio en la evolución natural de la enfermedad, existiendo hoy terapias orientadas a controlar las principales vías patogénicas involucradas: ON, PG, y ET. Los principales factores pronósticos que permiten guiar la terapia y la adición de fármacos específicos a la terapia inicial son: clase funcional, ECO, NT pro-BNP, distancia recorrida en el test de caminata de seis minutos y variables hemodinámicas del cateterismo. El Trasplante bi-pulmonar está reservado para los pacientes que no responden al tratamiento médico en asociación máxima para el medio en que le paciente se encuentre.
Pulmonary arterial hypertension (PAH) is a chronic disease, characterized by increased pulmonary vascular resistance (PVR) at the pulmonary arterioles, which causes a progressive overload and subsequent dysfunction of the right ventricle (RV), which final stages leading to right heart failure, which seals their prognosis. PAH is more common in young women in middle-age, with the median survival of 2-3 years, before the advent of targeted therapies. The genetic basis suggests an autosomal dominant inheritance with incomplete penetrance, mainly recognizing the condition of BMPR2. An alteration in the etiopathogenesis is recognized in the control signals mainly vasoconstrictor-vasodilator balance level endothelium, with an imbalance towards proliferation and vasoconstriction, which are involved three pathogenic pathways: The nitric oxide (NO), Prostacyclin (PG) and endothelin (ET). Early diagnosis of PAH is associated with better long-term survival, so the search for a patient with dyspnea, fatigue, chest pain and / or syncope, as well as at-risk populations, such as with relatives 1 PAH, scleroderma and Portal Hypertension carriers, should be the strategy of choice. Doppler (ECO) Echocardiography is the most widely used research tool in current clinical practice. The diagnosis should be confirmed by right heart catheterization, with direct measurement of pulmonary artery pressure, and vasoreactivity testing should be performed. The advent of drug-specific PAH treatment has caused a change in the natural history of the disease, existing today aimed at controlling major pathogenic pathways involved therapies: ON, PG, and ET. The main prognostic factors that help guide therapy and the addition of specific drugs to initial therapy are: functional class, ECO, NT pro-BNP, distance covered in the walk test six minutes and catheterization hemodynamic variables. The bi-lung transplant is reserved for patients who do not respond to medical treatment in full partnership for the environment in which patient you are.
- 1.
La hipertensión pulmonar (HAP), reconocida como el aumento de la resistencia vascular pulmonar a nivel de la arteriola pulmonar, fue descrita por Romberg en 1891. Sin embargo, solo desde la década del 90 con el avenimiento de las prostaciclinas existe una terapia específica que revoluciona el curso de la enfermedad, convirtiéndola en una entidad potencialmente tratable.
- 2.
Diagnóstico Precoz: La ecocardiografía efectuada por operadores experimentados es la herramienta de mayor utilidad de pesquisa ante la sospecha clínica de Hipertensión Pulmonar, así como en pacientes de grupos de riesgo familiares de pacientes con HAP, pacientes con Esclerodermia, entre otros.
- 3.
La confirmación diagnóstica debe realizarse en centros especializados mediante cateterismo cardiaco derecho y medición de Presión Arterial Pulmonar (PAP) Media, Presión de Enclavamiento o Capilar Pulmonar (PCP) y Gasto Cardiaco (GC).
- 4.
Durante el cateterismo se debe realizar el Test de Vasoreactividad Pulmonar para definir línea terapéutica a seguir. Y solo se usará Terapia con Bloqueadores de canales del Calcio en aquellos pacientes respondedores al test de vasoreactividad.
- 5.
En los pacientes con HAP no respondedores al test de vasoreactividad, se debe iniciar Terapia objetivo-específica, con acción en alguna de las vías patogénicas conocidas: Prostaciclinas, antagonistas de receptores de endotelinas, inhibidores de fosfodiesterasa, y estimuladores de la guanilato ciclasa soluble.
- 6.
En el seguimiento, la respuesta terapéutica será evaluada desde el punto de vista clínico, funcional y hemodinámico, con el propósito de definir la necesidad intensificación de la terapia.
- 7.
La derivación precoz al programa de trasplante bipulmonar es esencial en los casos en que el paciente se encuentre en clase funcional III-IV con terapia combinada óptima y no logre los objetivos de una buena respuesta.
- 8.
La única condición asociada a hipertensión pulmonar que puede tener una reversibilidad y prácticamente curación es la HAP asociada a tromboembolismo pulmonar crónico para la que existe la endarterectomía pulmonar, cirugía efectuada solo en centros especializados. Por lo que es fundamental descartar en entidad.
Se define Hipertensión Pulmonar (HP) como el aumento de la presión media de la arteria pulmonar (PAPm) ≥ 25mmHg. Para efectos de esta revisión nos referiremos principalmente a la hipertensión arterial pulmonar (HAP) del grupo I de la OMS (Tabla 1), antiguamente llamada “Primaria”, ya que esta es una condición que ha tenido un extraordinario avance en el desarrollo de nuevas alternativas terapéuticas1,2.
CLASIFICACIÓN HIPERTENSIÓN PULMONAR NIZA 2013
| 1. Hipertensión Arterial Pulmonar |
|---|
| 1.1 HAP idiopática |
| 1.2 HAP hereditaria (BMPR2, ALK-1, ENG, SMAD9, CAV1, KCNK3, desconocida) |
| 1.3 Inducidas por drogas y toxinas |
| 1.4 Asociada a: Enfermedad Tejido Conectivo, HIV, Hipertensión Portal, Cardiopatías Congénitas, Schistosomiasis |
| 1 Enfermedad veno oclusiva pulmonar, hemangiomatosis capilar pulmonar |
| 1 HTP persistente del recién nacido |
| 2. Hipertensión Pulmonar debido a Enfermedad Cardiaca izquierda |
| 2.1 Disfunción ventricular sistólica o diastólica, enfermedad valvular, cardiopatías con obstrucción del tracto de entrada o salida, cardiomiopatías congénitas. |
| 3. Hipertensión Pulmonar debido a Enfermedad Pulmonar y/o Hipoxia |
| 3.1 Enfermedad Pulmonar obstructiva crónica |
| 3.2 Enfermedad Intersticial Pulmonar |
| 3.3 Otras enfermedades con patrones obstructivos y restrictivos mixtos |
| 3.4 Síndrome de Apnea del Sueño |
| 3.5 Síndromes de hipo ventilación alveolar |
| 3.6 Exposición cónica a altitud |
| 3.7 Enfermedades Pulmonares del desarrollo |
| 4. Hipertensión Pulmonar por tromboembolismo pulmonar crónico (CTEPH) |
| 5. Hipertensión Pulmonar con mecanismo incierto o multifactorial |
| 5.1 Hematológicos: Anemia hemolítica crónica, Síndrome Mieloproliferativo, Esplenectomía |
| 5.2 Enfermedades Sistémicas: Sarcoidosis, Histiocitosis pulmonar, linfangioleiomiomatosis |
| 5.3 Enfermedades Metabólicas: Enfermedad Gaucher, Tiroideas |
| 5.4 Otras: Obstrucción tumoral, fibrosis mediastinica, Falla renal cronica, HTP segmentaria |
Traducida de 5th WSPH Nice 2013. Update Clinical Clasification Simonneau G.et al. J Am Coll Cardiol 2013:62(25), Suppl D.
La HAP es una enfermedad crónica y progresiva, de baja prevalencia, pero alto impacto por su elevada mortalidad. Se define la HAP del punto de vista hemodinámico invasivo, como el aumento de la PAPm ≥ 25mmHg, con una presión capilar pulmonar (PCP) ≤ 15mmHg1,2.
Se ha estimado para la HAP (fórmula de la National Institute of Health) un promedio de sobrevida de 2, 8 años, o una sobrevida promedio de 40% a dos años1,2. Múltiples son los mecanismos patogénicos involucrados que conducen a través de una progresiva obliteración del lumen vascular pulmonar, secundario a proliferación de la capa media e íntima, que lleva a un aumento progresivo de la resistencia vascular pulmonar (RVP) y luego al aumento de la presión arterial pulmonar (PAP) y falla cardiaca derecha. Existe un sustrato genético para esta condición, que se reconoce tendría el carácter de autosómico dominante con penetración incompleta, es decir solo el 25% de los portadores del gen desarrollan la enfermedad3.
La baja prevalencia y la ausencia de síntomas específicos es la razón por la cual la HAP permanece como una condición poco reconocida y sub-diagnosticada, siendo los síntomas más comunes: disnea, dolor torácico, fatiga y síncope1–4. Esta entidad afecta fundamentalmente a mujeres jóvenes (relación 8:1 con hombres), en plena edad productiva desde el punto de vista familiar, social y laboral, que en el momento del diagnostico la mayor parte se encuentra en Capacidad Funcional III, los hallazgos reportados en una serie nacional son similares a los reportados en la literatura5.
En Chile, se desconoce la real prevalencia de la enfermedad, sin embargo lo reportado en Francia y Reino Unido: Prevalencia de 15–25/millón de habitantes e incidencia de 1-2/millón de habitantes/año1,6,7.
En la actualidad existe consenso en que la terapia farmacológica mejora la sobrevida y calidad de vida de la HAP, con suficiente sustento en la literatura y que cuenta con la aprobación por la FDA y agencia regulatoria europea (EMA). Galie et al. muestra en su meta análisis que las terapias específicas han demostrado reducir la mortalidad del orden del 43%, cambiar el curso natural de la enfermedad y mejorar la calidad de vida, permitiendo el regreso de estos pacientes a una vida social, familiar y laboral activa2,8,9. A nivel nacional, un estudio en pacientes que tuvieron acceso a terapias especificas para HAP mostró una sobrevida de 82% a 3 años versus la esperada de 45%, de acuerdo a la fórmula del NIH10. Es por ello que creemos debe efectuarse el máximo esfuerzo por permitir que estos pacientes accedan a estas terapias, en la modalidad que requieran, según los riesgos inherentes y se evite la progresión de la enfermedad, en cuyo caso la única alternativa es el trasplante bipulmonar11,12.
CLASIFICACIÓNDesde el primer encuentro de expertos en Evian 1998, hasta el V encuentro mundial de Niza 20131, se ha complementado y perfeccionado la clasificación de HP estratificando en 5 grupos llamados de la OMS 1-V (Tabla 1). A continuación se detallan los principales dos grupos en los cuales se ha centrado la investigación orientada al reconocimiento de vías patogénicas susceptibles de ser intervenidas mediante terapias farmacológicas específicas: Grupo 1 y 413.
1-Hipertension Arterial Pulmonar (HAP):Este es el grupo al que se han orientado los principales avances en términos terapéuticos. Antiguamente se le denominó “primaria” y se subclasifica en: Idiopática, heredable o asociada a drogas (anorexígenos, anfetaminas), HIV, mesenquimopatías (lupus, esclerodermia, enfermedad mixta del tejido conectivo ente otras), hipertensión portal y cardiopatías congénitas con shunt intracardiaco (Eissenmenger). Este es el grupo al que se enfocan la mayor cantidad de nuevas terapias y es el objetivo principal de esta revisión.
4-HTP por Enfermedad Tromboembólica Crónica:Esta es la única condición potencialmente curable a través de una endarterectomía pulmonar, cirugía que debería ser efectuada en centros de experiencia por lo cual es fundamental descartar esta condición dentro del proceso de estudio de los pacientes con HP.
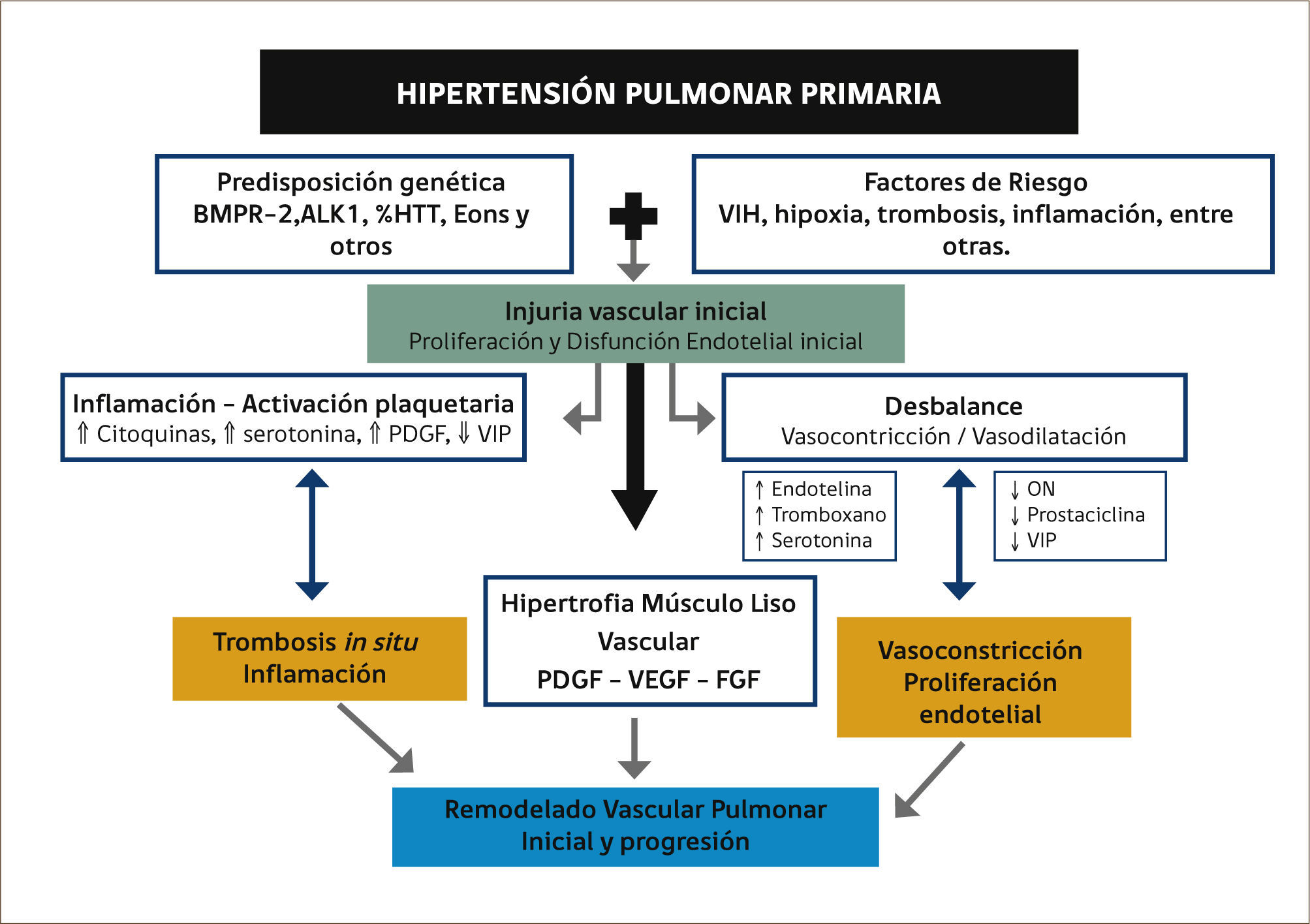
FISIOPATOLOGÍA HIPERTENSIÓN ARTERIAL PULMONARLa base fisiopatológica que subyace al aumento de la resistencia vascular pulmonar (RVP) es la enfermedad vascular hipertensiva en arterias de pequeño tamaño y arteriolas pulmonares. En su desarrollo participan múltiples factores celulares y moleculares que dan lugar al remodelado de la pared del vaso por cuatro mecanismos fundamentales, que son2,14:
- 1.
La vasoconstricción.
- 2.
La proliferación celular.
- 3.
La trombosis.
- 4.
Los factores inmunitarios / inflamatorios.
El origen es desconocido, pero se postula la existencia de una predisposición genética sobre la que deben actuar factores facilitadores y desencadenantes que dan lugar al inicio de la enfermedad3. Una base genética poligénica sobre la cual se superponen factores facilitadores y gatillantes, inflamatorios y ambientales que permiten el desarrollo y progresión de la enfermedad3.
Los diferentes mediadores moleculares implicados en el desarrollo de la enfermedad y los principales mecanismos de acción y el tipo celular implicado se ilustran en la Figura 1. El efecto final de estos mediadores es un desbalance hacia los que favorecen la vasoconstricción, inflamación, la proliferación celular y la trombosis vascular frente a los que ejercen el mecanismo contrario. Se ha relacionado una excesiva vasoconstricción con la función o expresión anómala de los canales de potasio en el músculo liso vascular, así como también con la disfunción endotelial. La disfunción endotelial está involucrada con una menor producción de agentes vasodilatadores como el óxido nítrico (ON) y prostaciclina (PG), junto con la mayor expresión de sustancias vasoconstrictoras y proliferativas como la Endo-telina (ET) y el Tromboxano A2 (TxA2). El conocimiento de estos mediadores no sólo es importante para entender la historia natural de la enfermedad, sino porque son las dianas a las que se dirigen los diferentes tratamientos actuales y las nuevas líneas de investigación. En la actualidad se reconocen tres vías patogénicas, que son además blancos terapéuticos: Vía ON, vía PG y la vía de la ET2,14.

En los últimos años han habido grandes avances en este campo, fundamentalmente en el estudio de los genes BMPR2 (gen del receptor tipo II de la proteína morfogenética ósea), ALK1 (activin-receptor-like kinase1) y 5HTT (endoglina asociada a la telangiectasia hemorrágica familiar y el gen del transportador de serotonina), cuyo polimorfismo LL (dos alelos largos) parece ser más frecuente en pacientes con HAP que en los controles3,14.
El remodelado vascular lleva a una obstrucción progresiva del lecho pulmonar, con el aumento de la RVP y de la post carga del ventrículo derecho (VD), que lleva a la hipertrofia y posterior dilatación del VD derecho, lo cual lleva finalmente al deterioro funcional y falla cardiaca derecha2,14.
DIAGNÓSTICOEl diagnóstico suele sospecharse en el contexto de paciente mujer joven, con síntomas vagos tales como disnea, fatiga, dolor torácico, síncopes, palpitaciones, entre otros. El diagnóstico de HAP es un diagnóstico de descarte, para lo cual se sugiere complementar el estudio inicial con una amplia batería de exámenes de laboratorio e imágenes. El Ecocardiograma es el principal examen de pesquisa, a pesar la variabilidad inter e intraobservador, la falta de correlación fina con el cateterismo y la ausencia de información cuando el reflujo tricuspídeo no es detectado. En manos expertas, una ecocardiografía que muestre una presión sistólica de arteria pulmonar (PAPS) >36mmHg con cavidades derechas dilatadas, sumado a un contexto clínico compatible obligan a efectuar un esfuerzo confirmatorio de esta condición dadas las repercusiones clínicas, psicológicas, laborales, económicas y éticas que involucra el diagnóstico de HAP. El examen confirmatorio mediante técnica invasiva, en laboratorio de hemodinamia es el cateterismo derecho con las siguientes cifras: PAPM ≥ 25mmHg en reposo con capilar pulmonar ≤ 15mmHg1,2.
ClínicaEn etapas iníciales la HAP puede ser asintomática. Cuando están presentes los síntomas son inespecíficos y a menudo son difíciles de diferenciar de otras enfermedades pulmonares o cardiovasculares. El intervalo entre el inicio de los síntomas hasta el diagnós-tico puede ir entre 2 y 3 años1,2,5. La disnea de ejercicio es el síntoma de presentación más frecuente, le siguen en importancia el dolor torácico, la fatiga y el síncope, en raras ocasiones, los pacientes pueden presentar hemoptisis. En Chile el estudio de Zagolin y cols, el 83% de los pacientes tenía dos o más síntomas en el momento de la consulta al centro de referencia5.
Un examen físico normal no descarta hipertensión pulmonar. Sin embargo, cuando presentan anormalidades del examen físico tienden a localizarse en el sistema cardiovascular. Un examen cuidadoso suele detectar signos de la HP y de hipertrofia VD. Los hallazgos más frecuentes presentes son: 1) el componente pulmonar acentuado del segundo ruido cardiaco (P2) se observa en 90% de los pacientes con HAP y 2) un soplo holosistólico de regurgitación tricuspídea. La distensión venosa yugular (onda a), la hepatomegalia, el edema periférico, la ascitis y las extremidades frías caracterizan a un paciente en un estado más avanzado1,2. Por lo general, la auscultación pulmonar es normal, sin embargo la auscultación de bruits (soplo pulmonar) sugiere descartar etiología tromboembólica crónica.
El diagnóstico de HAP es un diagnóstico de descarte de otras enfermedades que podrían ocasionar disnea, fatiga, dolor torácico o síncope, por lo que se propone la estrategia escalonada de diagnóstico. Figura 2.
Utilidad de la radiografía de tórax
La radiografía de tórax tiene un limitado beneficio, no obstante, en el 90% de los pacientes con HAP la radiografía torácica es anormal en el momento de realizar el diagnóstico1,2,15–18. Los resultados incluyen dilatación arterial pulmonar central, que contrasta con la «poda» (pérdida) de los vasos sanguíneos periféricos. La aurícula derecha y el aumento de tamaño del ventrículo derecho (VD) pueden darse en los casos más avanzados. Así también, la radiografía torácica permite excluir enfermedades pulmonares asociadas y la hipertensión venosa pulmonar causada por cardiopatía izquierda. En general, el grado de la HP en cualquier paciente no está en correlación con el de las anomalías radiográficas. Los signos radiológicos torácicos han sido evaluados para hipertensión pulmonar, encontrándose baja sensibilidad y buena especificidad13.
ElectrocardiogramaEl ECG puede proporcionar evidencias que indiquen o respalden la evidencia de HP: Desviación del eje QRS a derecha, ondas P prominentes en las derivaciones inferiores, onda R mayor que onda S en V1 – V2 (R/S 1) e hipertrofia del ventrículo derecho. No obstante, la ausencia de estas alteraciones no descarta la presencia de HTP1,2,15.
EcocardiografíaEs la herramienta de mayor utilidad en la sospecha, o screening de Hipertensión pulmonar, cabe destacar que es dependiente de la experiencia del operador y de la presencia de reflujo tricuspideo. En la actualidad es una herramienta ampliamente disponible, siendo su sensibilidad y especificidad del 69% y 94% respectivamente, en el diagnóstico de HAP, lo que implica que en un porcentaje de pacientes se puede subestimar o sobreestimar la PAPm1,2,19–22. Por otro lado, la ecocardiografía nos permitirá descartar o confirmar patologías cardiacas, así como cardiopatías congénitas que evolucionan con HTPulmonar y la insuficiencia cardiaca por enfermedades del músculo cardiaco, valvular, o isquémicas1,2. En la Tabla 2 se indica según los parámetros de la ecocardiografía y la probabilidad de HP.
ECOCARDIOGRAFÍA COMO PESQUISA DE HAP
| DIAGNÓSTICO | VELOCIDAD IT Y PAPS | VARIABLES ADICIONALES AL ECO |
|---|---|---|
| Improbable | Velocidad IT 2,8 mt/seg y PAPS 36 mmHg | Ausentes |
| Posible | Velocidad IT 2,8 mt/seg y PAPS 36 mmHg | Presentes |
| Posible | Velocidad IT 2,9 a 3,4 mt/seg y PAPS 37-50 mmHg | Si |
| Probable | Velocidad IT 3,4 mt/seg y PAPS 50 mmHg | 4 |
Los hallazgos sugerentes de HAP son los siguientes:
- 1.
- PAPS 50mmHg, o
- 2.
- PAPS entre 36-50mmHg con cavidades derechas dilatadas o signos de insuficiencia VD, asociado o no a derrame pericárdico.
Se sugiere o recomienda que todo paciente con clínica sugerente, y ecocardiografía compatible, habiendo descartado razonablemente otras posibilidades diagnósticas, debe ser derivado a un centro de referencia para confirmar o descartar el diagnóstico de HAP1,2. En la actualidad existe consenso que es fundamental efectuar un cateterismo cardiaco derecho mediante catéter de Swan Ganz para confirmar el diagnóstico, evaluar la respuesta vasodilatadora, que da cuenta de la reactividad vascular del territorio vascular y susceptible de responder a terapia con bloqueadores del calcio1,2.
En pacientes asintomáticos con esclerodermia se recomienda realizar un ecocardiograma anual buscando hipertensión pulmonar1,2. En otras enfermedades del colágeno se recomienda la ecocardiografía sólo si hay síntomas1,2. Cabe destacar que si bien la ECO es el principal método de pesquisa, no es el más idóneo ya que es tardío, debido a que cuando existe HP a nivel ecográfico, más del 50% del lecho vascular pulmonar ya se encuentra afectado.
El estudio con Angio TAC Pulmonar permite el diagnóstico diferencial con enfermedad tromboembólica pulmonar y otras pato- logías pulmonares que pueden evolucionar con hipertensión pulmonar. Una arteria pulmonar (AP) 29mm tiene una sensi- bilidad del 87% y una especificidad del 89% para diagnóstico de HAP1,2. El AngioTAC no sustituye al Cintigrama pulmonar de ventilación perfusión en el proceso de descarte de enfermedad tromboembólica crónica (CTEPH), teniendo este último un mejor valor predictivo negativo del orden de 99%1,2.
Dentro del proceso diagnóstico y diagnóstico diferencial con múltiples patologías que pueden evolucionar con hipertensión pulmonar deben efectuarse pruebas de laboratorio clínico (hemograma, pruebas hepáticas, función renal y tiroidea, entre otras), pruebas de función pulmonar (espirometría, difusión CO), gases arteriales, serologías virales como VIH, auto anticuerpos ante la sospecha de una enfermedad del mesenquima, polisomnografía en síndrome de apnea del sueño, etc. Por otro lado, se debe realizar pruebas tendientes a objetivar la capacidad funcional, entre ellas la prueba de caminata de 6 minutos y la prueba cardiopulmonar, así mismo deberá contar con la medición de NT pro BNP1,2.
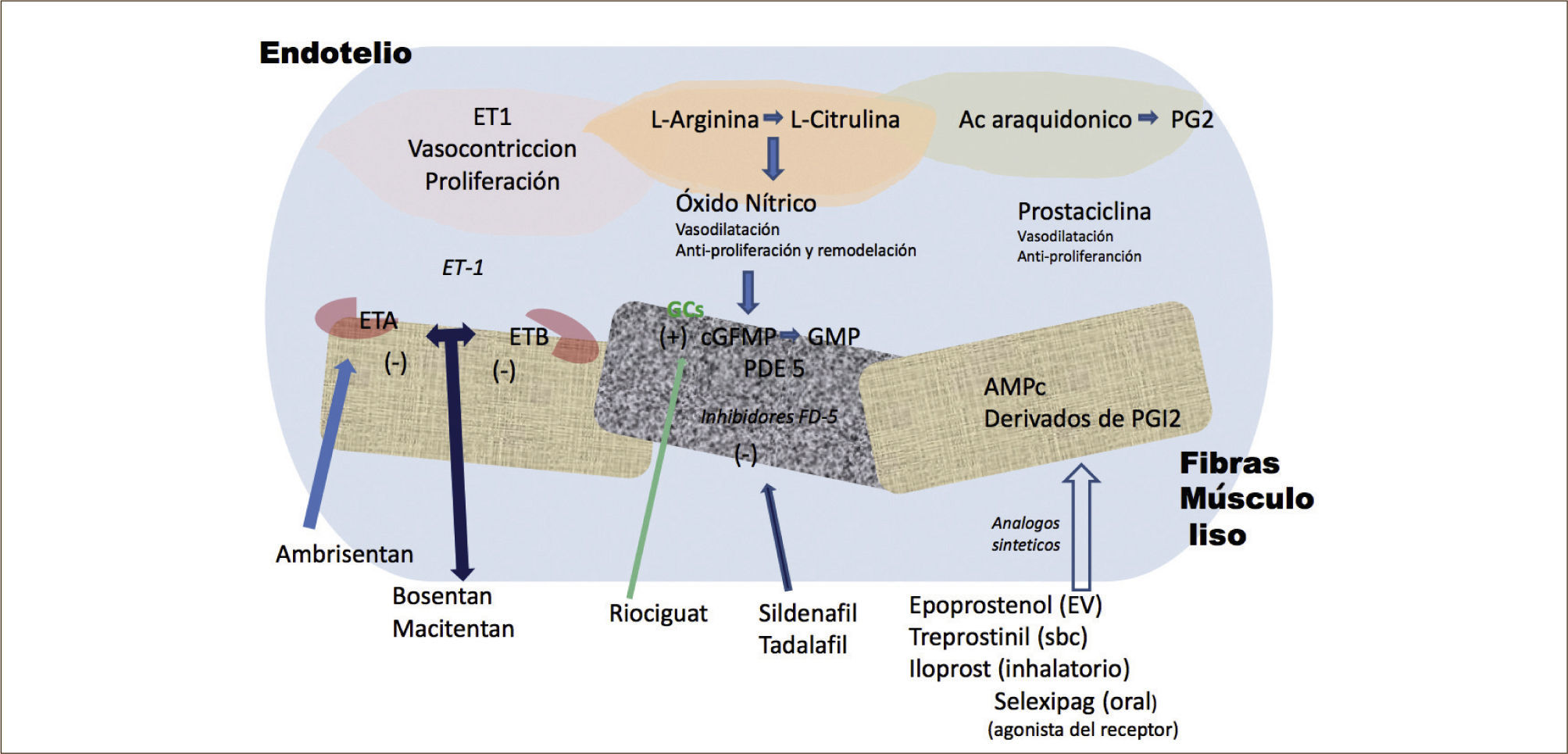
TRATAMIENTOEn la etiopatogenia de la HAP juega un rol preponderante la disfunción del endotelio, pérdida de los mecanismos normales de vasodilatación, entre los que participa el óxido nítrico y las prostaciclinas y aumento del efecto vasoconstrictor de diversos mediadores como endotelinas, tromboxano2,14. En la actualidad las terapias en HAP son “objetivo-específicas”, es decir están dirigidas a algunas de la vías patogénicas conocidas, ya sea de la vía Prostaciclina, del óxido nítrico (ON), o endotelinas como se comenta a continuación2,14,23,24. Figura 3
- I.
En la vía de las prostaciclinas; Epoprostenol (prostaglandina endovenosa), Iloprost o treprostinil (prostaglandina nebulizada), treprostinil (prostaglandina subcutánea), treprostinil, y selexipag (análogo no prostanoide oral).
- II.
En la vía del ON: Los inhibidores de fosfodiesterasas como Sildenafil-tadalafil (inhibidor fosfodiesterasa 5); y los recientemente aprobados estimuladores de la guanilato ciclasa Riociguat.
- III.
En la vía de los antagonistas de receptores de endotelina como Bosentan-Ambrisentan-Macitentan (ERAS).

En la actualidad existe consenso que previo al inicio de cualquier terapia, probada o experimentales, fundamental efectuar un estudio hemodinámico mediante catéter de Swan Ganz, que junto con confirmar el diagnóstico, evalúa la respuesta vasodilatadora que da cuenta de la reactividad vascular del territorio vascular remanente, y susceptible de responder a terapia1,2,23. La prueba de vasoreactividad puede realizarse con óxido nítrico, adenosina o prostaglandinas2,14,23,24. La prevalencia de vasoreactividad en la literatura es de 12% según el estudio francés de Sitbon y col.25 y en la experiencia nacional es muy similar, de 11%26. Solo los pacientes vasoreactivos se benefician del uso de bloqueadores del calcio (BBC) tipo diltiazem, o dihidropiridinas como: Amlodipino o Nifedipino2,23. En aquellos en los que no se logra una respuesta vasodilatadora adecuada (no vasoreactivos), se benefician de las terapias objetivo-específicas disponibles, de demostrada eficacia que involucran las principales vías patogénicas de la HAP2,23.
La terapia con prostaciclinas endovenosa es la mejor terapia disponible ya que ha demostrado mejorar sobrevida, calidad de vida, variables hemodinámicas, Capacidad Funcional y la distancia recorrida, sin embargo su uso es complejo y muy costoso2,8,23. En Chile, en la actualidad, se encuentran disponibles en la via del ON: Sildenafil-Tadalafil oral y Riociguat. En la vía de la prostaciclinas: tanto nebulizada como subcutánea, Iloprost y Treprostinil respectivamente. En la vía ET: Bosentan, de uso oral. En 2015 se ha programado la autorización por la agencia regulatoria de ambrisentan, macitentan, selexipag y treprostinil oral.
En los pacientes en CF II se puede iniciar la terapia con una droga oral (Monoterapia), Sildenafil ha demostrado ser efectivo en el tratamiento de pacientes con HAP2,23 y es la droga más frecuente utilizada en la terapia mayor disponibilidad y costo en nuestro país. En la Tabla 3 se muestran otras alternativas de inicio drogas como Monoterapia de HAP en CF II.
TERAPIA FARMACOLÓGICA EN HAP
| TERAPIAS EN CLASE FUNCIONAL II | TERAPIAS EN CLASE FUNCIONAL III | TERAPIAS EN CLASE FUNCIONAL IV |
|---|---|---|
| • Sildenafil / Tadalafil | • Sildenafil / Tadalafil | • Epoprostenol endovenoso en bomba infusión continua |
| • Bosentan / macitentan | • Bosentan / macitentan | |
| • Ambrisentan | • Ambrisentan | |
| • Lloprost nebulizado | • Iloprost nebulizado, o treprostinil vo o nebulizado, o selexipag | |
| • Riociguat | • Riociguat | |
| • Iloprost ev | Sildenafil / Tadalafil | |
| • Treprostinil ev | Bosentan / macitentan | |
| Ambrisentan | ||
| Treprostinil, iloprost, selexipag | ||
| Riociguat | ||
| Solos o en combinación de | ||
| 2.3 drogas | ||
| INICIO COMO MONOTERAPIA | TERAPIA COMBINADA INICIAL (2 drogas) | TERAPIA COMBINADA INICIAL (Doble o Triple) |
Se recomienda en las guías de Niza 201323, que los pacientes no respondedores a la monoterapia inicial luego de tres meses, escalen a terapia de combinación (biasociada) y si luego de otros tres meses no hay respuesta satisfactoria, se proceda a terapia triasociada, e incorporación al programa de trasplante bipulmonar8,12,13. Esta modalidad de terapia escalonada según metas es lo que se ha denominado “terapia guiada por objetivos”. Los parámetros que se evalúan para considerar respuesta son: clase funcional de la OMS, distancia recorrida en caminata de 6min, presión arterial sistólica de la arteria pulmonar (PAPS por Eco), presencia de derrame pericardico y excursión sistólica antero – posterior del anillo tricuspideo (TAPSE) en ecocardiografía, consumo de oxígeno peak en Test Cardiopulmonar cuando se dispone de el, BNP o Pro-BNP y algunos parámetros hemodinámicos cuando se repite el cateterismo tales como PAPm, presión de aurícula derecha (PAD).
Se debe considerar que estas drogas específicas pueden asociarse unas con otras, sin ajuste de dosis, ya que no se ha observado una potenciación de efectos adversos2,23. La única excepción la constituye la asociación de Sildenafil con Riociguat por aumento del efecto hipotensor27.
La base de la terapia escalonada y guiada por objetivos se ha basado en los trabajos hasta el momento disponibles sin embargo en 2014 se ha presentado en el Congreso Europeo de Munich, el estudio Ambition28, pronto a ser publicado, en que se demuestra que el enfrentamiento inicial con dos drogas (ambrisentan-tadalafil) es superior a la aproximación de cada una de ellas por separado y el grupo francés ha demostrado en un reporte de 18 pacientes jóvenes con HAP severa que la aproximación con triple terapia desde el inicio (epoprostenol ev, bosentan y sildenafil) logra una estabilización realmente significativa sin precedente29 de manera que es posible que en el futuro próximo algunos paradigmas en el tratamiento se modifiquen y sea la terapia asociada ya sea doble o triple inicial la que lidere nuestra aproximación terapéutica inicial.
Terapia CombinadaExisten numerosos ensayos que demuestran que la combinación de estas sustancias es segura y efectiva2,23,28,29. Un reciente meta-análisis de 6 ensayos randomizados controlados con terapia combinada, que incluyeron 858 pacientes comparó con grupo control y terapia combinada, redujo el riesgo de deterioro clínico en manera significativa (RR 0.48;95% IC:0.26-0.91;p=0.023), incrementó la distancia recorrida en 22 metros y redujo la PAPm, la presión de aurícula derecha, así como la resistencia vascular pulmonar. La incidencia de eventos adversos fue similar en ambos grupos9.
Se sugiere la terapia combinada en clase funcional de la OMS III y IV en los casos en que de acuerdo a la aproximación. “Terapia guiada por objetivos”, sugerida en las guías europeas 20092 y reforzada en el último simposio de expertos Niza 201323,24, no se logran los objetivos que sugieren buena respuesta al tercer mes de monoterapia Figura 4.

Finalmente, es necesario enfatizar en el hecho que una vez que el paciente entra en la categoría de terapia combinada y logra estabilización con buenos parámetros según la aproximación de “terapia guiada por objetivos”, ésta no se debe suspender y deberá ser sostenida permanentemente a menos que presentare algún evento adverso o existiera otra combinación aprobada por los mecanismo regulatorios más beneficiosa.
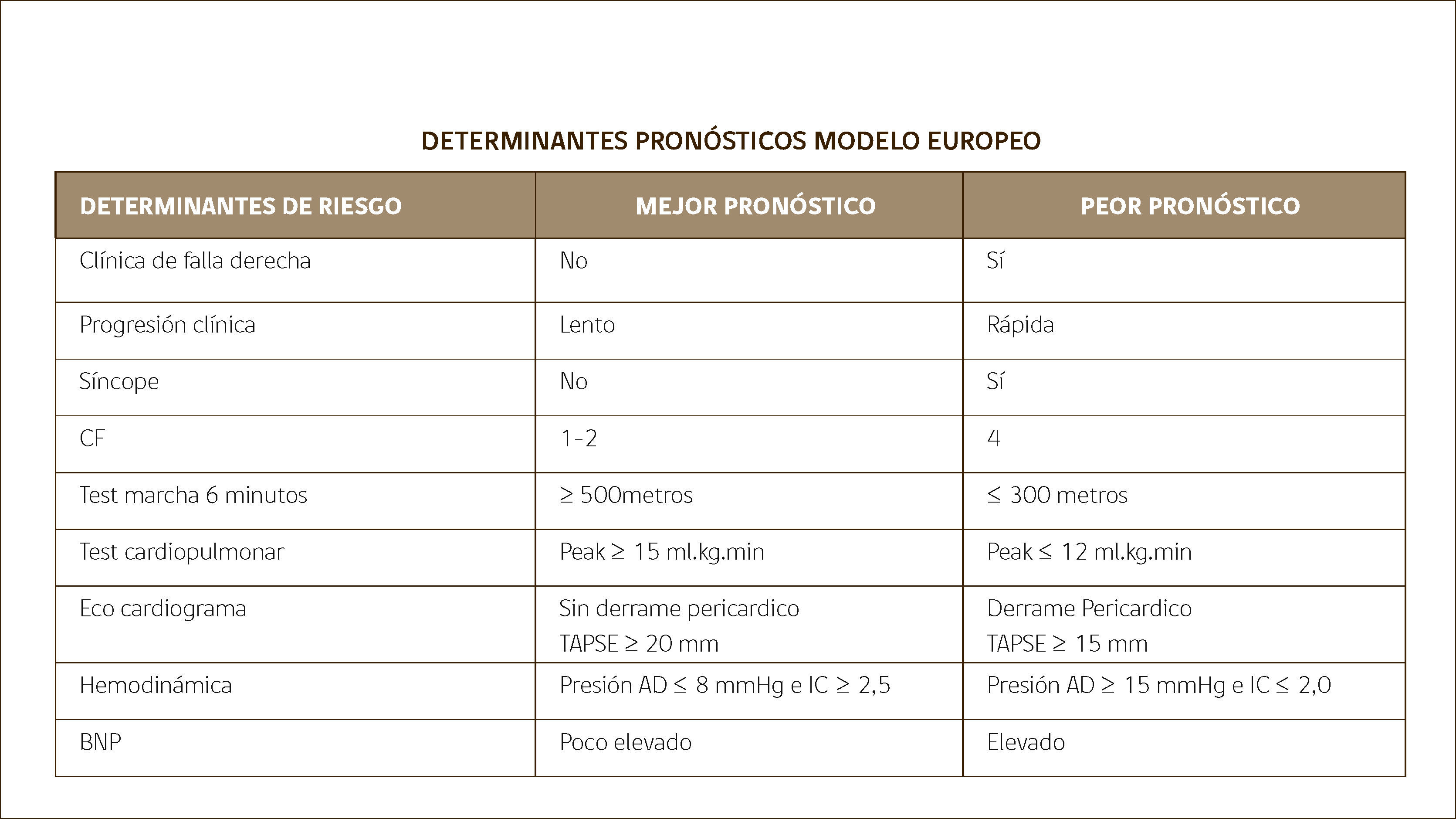
Seguimiento y Factores Pronósticos en HAP (Figura 4)El seguimiento de los pacientes con HAP es clínico, funcional y hemodinámico. Se recomienda que los pacientes sean evaluados luego del diagnóstico definitivo, en un centro de referencia en los meses: 1-3-6-12 y luego del primer año, si se mantiene estable, cada seis meses.
- a)
Clínico: Basado en Clase Funcional de la OMS (I a IV) asociado a una evaluación clínica dirigida a la búsqueda de elementos de disfunción ventricular derecha, eventos adversos, nivel funcional, calidad de vida y estado funcional laboral, social y familiar.
- b)
Laboratorio: Se sugiere el uso de biomarcadores tales como Pro-BNP o BNP o Nt-ProBNP como estimación del nivel de dilatación del ventrículo derecho. Este examen se recomienda esté disponible en cada control, ya que su nivel marca conducta. Es un marcador de respuesta favorable a la terapia, cuando sus niveles se acercan a la normalidad, o disminuye de un control a otro en más de un de 50% para considerar una respuesta favorable (Niza).
- c)
Funcional: Se sugiere en cada control disponer de la información aportada por la prueba de caminata de 6min, en el que lo más relevante es: la distancia recorrida y la presión arterial sistólica post ejercicio. Son elementos de mal pronóstico en el seguimiento de un paciente que ha iniciado terapia, la presencia de una distancia recorrida inferior a 400 mt.
- d)
Ecocardiografía: Se sugiere una nueva ecocardiografía de control al menos una vez al año en pacientes estables con buena respuesta clínica, funcional y hemodinámica. La ecocardiografía permitirá precisar dos elementos pronósticos relevantes para orientar la terapia del pacientes: La presencia o no de derrame pericárdico y el TAPSE como estimación sustituta de la funcionalidad del ventrículo derecho. En los casos de respuesta no satisfactoria a la terapia una monitorización más frecuente con ecocardiografía, como herramienta de evaluación de los cambios en la terapia.
Son candidatos a trasplante bipulmonar los pacientes en clase funcional III (en terapia combinada máxima disponible) o IV, y con enfermedad rápidamente progresiva11,12.
En la actualidad, la sobrevida con terapias farmacológicas especificas para HAP es superior al trasplante bipulmonar, de manera que es preferible agotar la instancia farmacológica antes de proceder a éste. Sin embargo, se sugiere enlistar ya en clase funcional III y terapia combinada, cuando el paciente no logra los objetivos enunciados en “terapia basada en objetivos”2,8,9,11,12,23,24.
En consecuencia y según lo reportado por la Sociedad Internacional de Trasplante de Corazón y Pulmón (www.ishlt.org) menos del 5% de todos los trasplantes Bi-pulmonares tienen como etiología la hipertensión pulmonar28. Cabe señalar que esta indicación va en disminución dada le mejoría de los resultados de las nuevas terapias de la HAP2,8,9,23,24. Sin embargo es importante no retrasar la incorporación del paciente a un programa de trasplante, cuando se encuentra en terapia combinada y capacidad funcional de la OMS avanzada (III), dado que la severidad de la insuficiencia ventricular derecha ensombrece la sobrevida post trasplante2,11,12,27,28.
Por otro lado, se debe considerar la etiología de la Hipertensión Pulmonar, dado que los pronósticos varían según ella; por ejemplo, los pacientes con Síndrome de Eisenmenger tienen mejor sobrevida que los pacientes con esclerodermia2. En el grupo con cardiopatía congénita se prefiere corregir el defecto e intentar bipulmonar por sobre el bloque corazón-pulmón usado en sus inicios. El subgrupo de peor pronóstico, cuya única terapia aprobada es el trasplante bi pulmonar, es la enfermedad veno-oclusiva y la hemangiomatosis capilar pulmonar2,11,12. En pacientes con HAP Porto Pulmonar la indicación es de Trasplante Hepático cuando la PAPm es menor 35mmHg ya sea a nivel basal o luego de terapia farmacológica29.
La sobrevida global post trasplante pulmonar a 5 años es de 45-50% y en promedio, 8 años23,28. Algunos reportes actuales señalan mejor sobrevida del orden de 45% - 66% a 10 años28,30. Se prefiere el trasplante bipulmonar basado en la evidencia de la International Society Heart Lung Transplantation (ISHLT)11,31–33. Se considera elegible para trasplante aquel paciente que no responde adecuadamente a la terapia máxima farmacológica disponible y tolerable. De acuerdo a lo anterior se han establecido criterios para ingresar a un programa de Trasplante12:
- -
Pacientes en clase funcional IV o en clase funcional III con máxima terapia disponible (terapia combinada) sin logro de objetivos de éxito.
- -
Pacientes con enfermedad veno-oclusiva o hemangiomatosis capilar pulmonar.
En casos muy seleccionados, en pacientes en CF OMS IV, pudiese considerarse la realización de una septostomia atrial de descarga del VD, como puente al trasplante, o como alivio sintomático en pacientes con insuficiencia cardiaca severa2.
TERAPIA NO FARMACOLÓGICAJunto con la terapia farmacológica específica, se considera de crucial importancia la adherencia a medidas no farmacológicas específicas que permiten de manera coadyuvante, beneficiar en la mejor calidad de vida y sobrevida de los pacientes. Se enumeran a continuación las medidas más relevantes que deben ser implementadas en estos pacientes:
RehabilitaciónLos pacientes con HAP deben ingresar a un programa de Rehabilitación cardiorrespiratoria y muscular (se recomienda sea supervisada en los primeros 3 meses, controlada con oximetría de pulso y frecuencia cardíaca y si saturación O2 es 91% se debe adicionar oxígeno por naricera 3 lt y continuar la rehabilitación luego de una pausa, y si la frecuencia cardíaca es 120/min se debe detener hasta su recuperación. Los ejercicios aprobados son en modalidad aeróbica (cardio) más resistivos y cargas livianas (1-2kg) asociado a tono, ejercicios respiratorios y equilibrio. Una vez completada la fase supervisada los pacientes pueden continuar su rehabilitación en domicilio (3-5 veces a la semana)2.
Terapia anticoagulanteEs un hecho reconocido que los fenómenos trombóticos por anormalidades en las vías de coagulación y fibrinólisis están presentes en la etiopatogenia y progresión de la enfermedad3,4. Se recomienda anticoagulación oral con INR 2-3 en HAP grupo I especialmente en el grupo idiopático2. No existe suficiente evidencia para su recomendación en HAP asociado a enfermedad del tejido conectivo. No se recomienda en HAP-portopulmonar. Se sugiere cautela en el grupo con cardiopatía congénita en atención a posibles comunicaciones bronquiales anómalas. No existe suficiente experiencia en este grupo con los nuevos anticoagulantes orales.
Terapia anticonceptivaEl embarazo se encuentra contraindicado en las pacientes con HAP, ya que el riesgo de muerte materna se encuentra entre el 30-40%6. Se recomienda el uso de métodos anticonceptivos tales como progestágenos IM, vo, de depósito, anillos vaginales, Mirena, o esterilización2. No se recomienda el uso de estrógenos en ninguna modalidad.
DiuréticosSuelen adicionarse diuréticos como furosemida o hidroclorotiazida asociados o no a medicamentos antialdosterónicos tipo espironolactona fundamentalmente en pacientes con evidencias de VEC (volumen extravascular aumentado) y en pacientes con evidencias clínicas o de laboratorio de insuficiencia ventricular derecha2.
OxigenoterapiaSe recomienda oxigenoterapia ambulatoria si la saturación basal es inferior a 91% con el propósito de mantener PO2 60mmHg la mayor parte del tiempo, 15 horas al día al menos2.
Apoyo psicológicoComo muchas enfermedades crónicas, la HAP sobre todo al momento del diagnóstico produce una ansiedad y aislamiento social significativo, por lo que se recomienda que los pacientes asistan a terapia de grupo o personalizada para lograr el apoyo necesario para sobrellevar su condición2.
Medidas GeneralesSe recomienda que los pacientes eviten altura en niveles sobre 1500 m (2l) y si deben efectuar viajes en avión, deben ser con oxígeno 2 o más litros por naricera previa autorización de su médico tratante. Se recomienda evitar drogas que definitivamente o probablemente puedan provocar HAP tales como aminorex, fenfluoramina, dexfenfloramina, cocaína, hierba de San Juan y fenilpropanolamina2. Los autores declaran no tener conflictos de interés, en relación a este artículo.