



Uno de los focos principales de la neurociencia es entender las funciones cerebrales superiores y cómo estas son inhibidas de manera reversible por los anestésicos generales. Por ello, la comprensión del mecanismo de acción de estos fármacos, que rutinariamente se utilizan en la práctica clínica, ha permitido avanzar enormemente en entender cómo se integra la información a nivel cerebral para establecer las nuevas memorias, la capacidad de reaccionar al medio externo y las bases de la consciencia. Desde la primera demostración pública exitosa del efecto de los anestésicos generales hasta nuestros días se ha determinado que los anestésicos actúan en bolsillos hidrofóbicos de receptores proteicos localizados en la membrana plasmática de las neuronas corticales y subcorticales, generando una disminución de la excitabilidad de las redes neuronales. Este efecto puede ser evidenciado por registros electroencefalográficos que han permitido registrar en línea el efecto de cada uno de los anestésicos. De este modo, se ha podido establecer que la anestesia es diferente al sueño, más bien corresponde a un coma farmacológico reversible. Sin embargo, pese a todos los avances, aún quedan muchas preguntas por responder, lo cual es el objetivo de las futuras investigaciones.
One of the main focus of neuroscience is to understand the higher brain functions, which are reversibly inhibited by general anesthetics. Therefore, understanding the mechanism of action of these drugs, which are routinely used in clinical practice, has allowed a great advance in how information is integrated in the brain to establish new memories, the ability to react to external environment and the consciousness. Since the first successful public demonstration of the effect of general anesthetics to the present day, it has been determined that anesthetics act on hydrophobic pockets of protein receptors in cortical and subcortical neurons, generating a decrease in the excitability of neural networks. This effect can be evidenced by electroencephalographic records that have allowed to register on line and to characterize the effects of each anesthetics. In this way, it has been possible to determine that anesthesia is different to sleep and rather corresponds to a reversible pharmacological coma. However, despite all of new evidence, there are still many questions to be answered, which is the goal of future researches.
La neurociencia estudia la estructura, la función, el desarrollo, la biología molecular, la farmacología y la patología del sistema nervioso central. Todo esto en su conjunto nos ha permitido comprender las bases de la memoria, las emociones, las conductas, la cognición, la consciencia, entre otras funciones cerebrales superiores. No obstante, aún existe un gran desconocimiento sobre estas mismas. Nuestra práctica anestésica nos evidencia día a día que las estructuras, mecanismos y redes neuronales, que dan cuenta de la memoria, movilidad, consciencia y nocicepción, son dependientes de blancos moleculares específicos sobre los cuales actúan los anestésicos generales1–3. Aún más, actualmente se ha observado que los efectos de los fármacos hipnóticos se han asociado a cambios en la cognición, tanto transitorios como persistentes en pacientes vulnerables luego de haber sido anestesiados4,5. Entonces, sin duda alguna el conocer los mecanismos moleculares de los anestésicos generales y como estos afectan las redes neuronales, ha permitido y permitirán profundizar en el conocimiento neurocientífico de las funciones cerebrales superiores de los seres vivos. En esta revisión se detallará el estado actual del conocimiento sobre los mecanismos de la anestesia general, destacando a los blancos moleculares y cómo éstos afectan a las redes cerebrales para producir los diferentes efectos clínicos observados en una anestesia general.
DEFINICIÓN DE ANESTESIA GENERALPara comenzar es conveniente definir el concepto de anestesia general (AG), pese a que es una tarea difícil y hasta la fecha no existe un definición consensuada. Operacionalmente, se considera que un paciente se encuentra bajo un estado de AG si tiene ciertas características. Entonces, utilizando dichas características contextualizaremos a que nos estamos refiriendo cuando hablamos de AG6.
Primero, debe existir un estado de inconsciencia, el cual debe ser generado y mantenido farmacológicamente y debe ser reversible al término de la anestesia.
Segundo, el paciente debe tener la imposibilidad de establecer recuerdos durante la cirugía, o sea, debe tener amnesia, la cual también debe ser reversible tras la anestesia.
Tercero, la inmovilidad es una condición que debe estar presente en una AG para que el acto quirúrgico se pueda realizar. Ciertamente el mecanismo por el cual se genera esta inmovilidad ha ido cambiando en el tiempo, desde la utilización de un sólo fármaco, como el éter, hasta el estado actual de una AG balanceada donde se utilizan bloqueadores neuromusculares para obtenerla. Finalmente, sin ser una condición absolutamente necesaria pero, totalmente deseable, en una AG se establece una inhibición de la nocicepción y al igual que la inmovilidad, el mecanismo como se establece esta inhibición ha ido cambiando en el tiempo. En suma, AG se podría definir como un estado reversible inducido farmacológicamente que se caracteriza por inconsciencia, amnesia, inmovilidad e inhibición de la nocicepción. Dado que es un estado similar a un coma, esta definición se podría simplificar en decir que la AG corresponde a un coma farmacológico reversible asociado a una mantención de la homeostasia interna3. Este coma farmacológico puede ser establecido con un único fármaco anestésico o un anestésico asociado a un bloqueador neuromuscular y a un opioide. El uso de un sólo fármaco implica la utilización de dosis elevadas que se asocian a efectos adversos, por ello actualmente se utiliza la combinación balanceada de fármacos, aprovechándose de la sinergia que se genera entre ellos (Figura 1). Con esto se logra disminuir las dosis y por ende los efectos adversos de éstos. A continuación, se abordará el tema de como mecanísticamente se obtiene el estado de una AG, considerando la perspectiva histórica y las particularidades de los principales fármacos anestésicos, tanto inhalatorios como intravenosos. Lo primero destacable de estos fármacos es que pese a las diferencias estructurales y fisicoquímicas, los anestésicos inhalatorios e intravenosos son capaces de producir el mismo estado1,7,8. Sin embargo, estas diferencias estructurales y fisicoquímicas permiten explicar por qué los fármacos intravenosos requieren concentraciones 10 veces menores que los fármacos inhalatorios2. Además, los anestésicos se caracterizan por establecer secuencialmente los diferentes efectos clínicos al ir incrementando las dosis, primero a bajas dosis se obtiene la amnesia, luego la inconsciencia y finalmente con dosis más elevadas se obtiene la inmovilidad (Figura 2)8–10. Por último, las curvas dosis respuesta poblacionales de los anestésicos generales se destacan por lo abrupto del establecimiento de los diferentes estados, lo que nos indica que los mecanismos sobre las funciones cerebrales superiores son altamente conservados. Teniendo en cuenta estas consideraciones generales, comenzaremos esta revisión con la perspectiva histórica del estudio de los mecanismos de la AG.
 en el caso de la inmovilidad y con los opioides para la inconsciencia. Además, la anestesia general per se contribuye a la analgesia generada por los opioides debido al establecimiento del estado de pérdida de consciencia. Todos estos efectos se producen con una mantención de la homeostasis interna.")
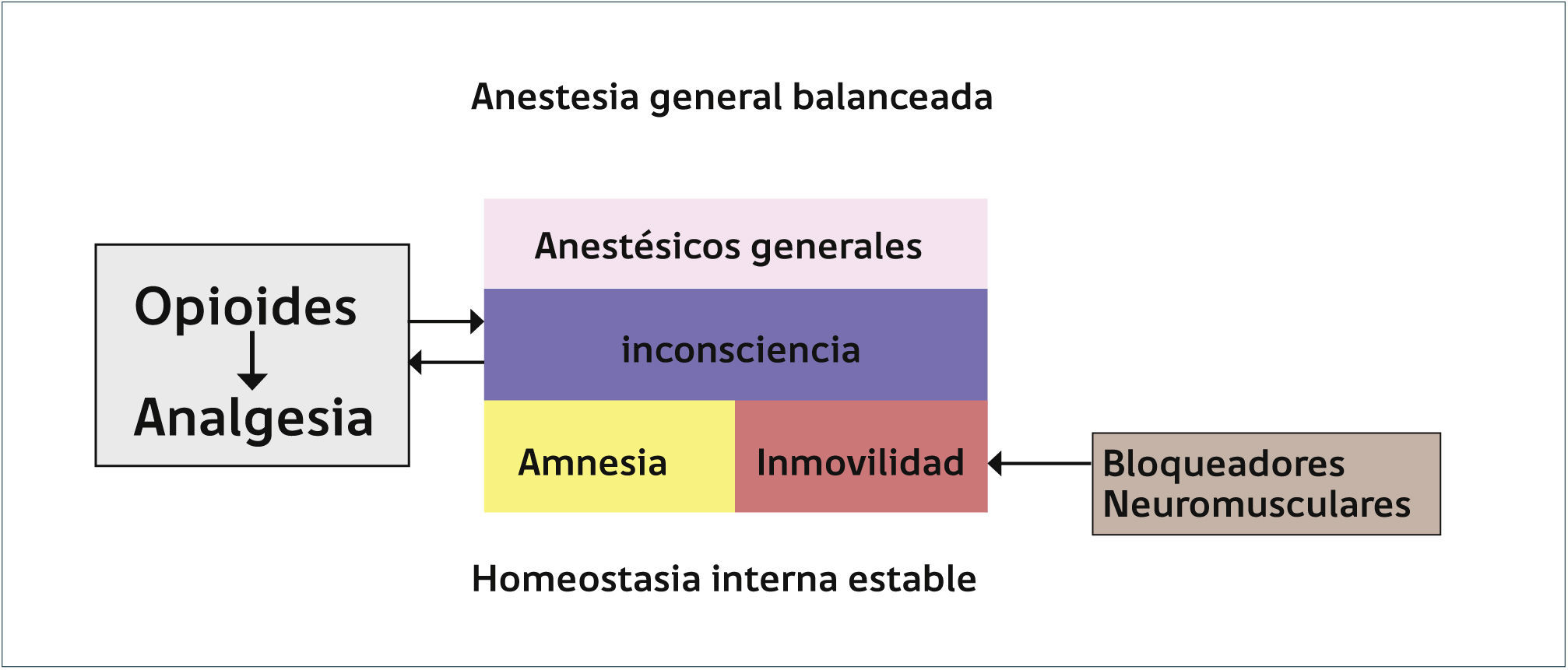
ESQUEMA RESUMEN DEL EFECTO DE LOS ANESTÉSICOS GENERALES EN UNA ANESTESIA GENERAL BALANCEADA
Los principales efectos de los anestésicos generales son la inconsciencia, amnesia e inmovilidad. Estos son complementados con los bloquedadores neuromusculares (BNM) en el caso de la inmovilidad y con los opioides para la inconsciencia. Además, la anestesia general per se contribuye a la analgesia generada por los opioides debido al establecimiento del estado de pérdida de consciencia. Todos estos efectos se producen con una mantención de la homeostasis interna.
..")
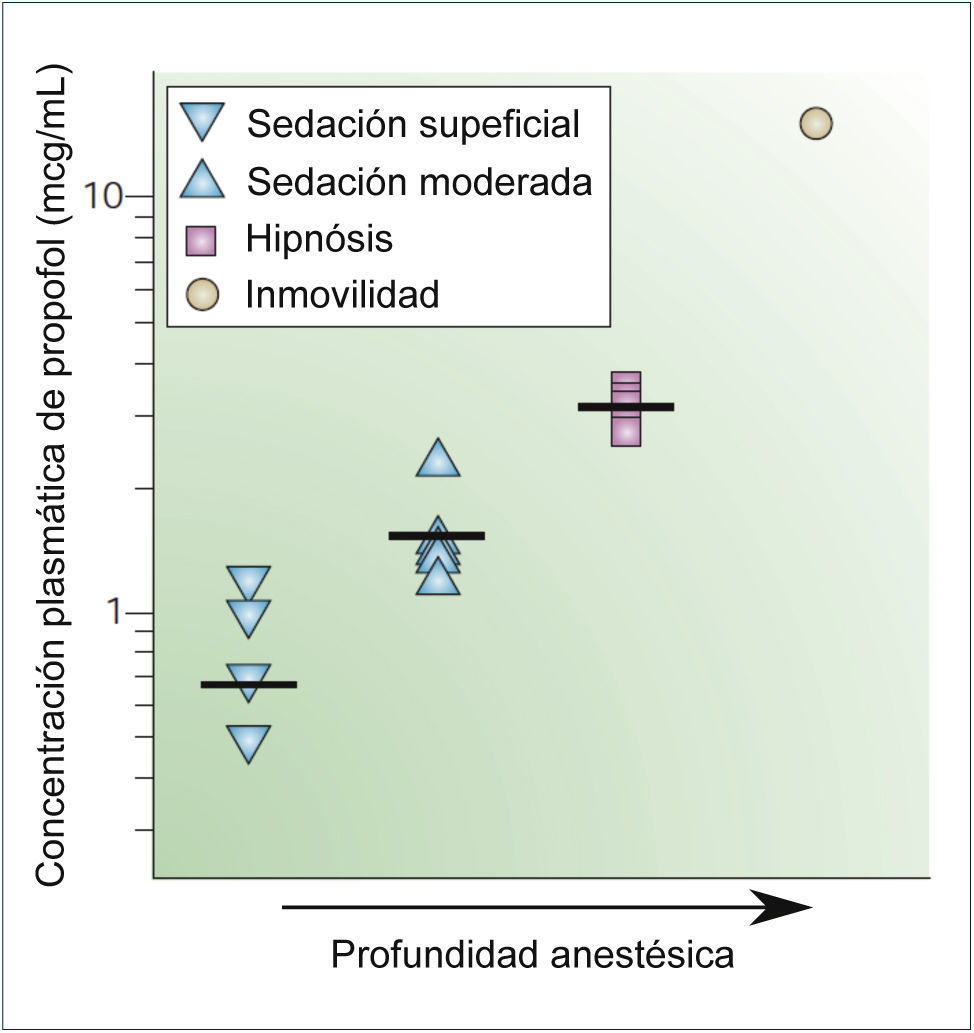
CORRELACIÓN ENTRE LAS CONCENTRACIONES PLASMÁTICAS DE PROPOFOL Y LOS EFECTOS ANESTÉSICOS
Los símbolos indican los valores obtenidos en diferentes estudios para establecer una sedación leve, sedación moderada, hipnosis e inmovilización. Las barras horizontales representan el promedio. Tomado y modificado de Rudolph, U. & Antkowiak, B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci 5, 709-720 (2004)..
Tras la primera demostración exitosa del efecto anestésico del éter en el año 1846 realizada por William Morton, en el lugar que actualmente es conocido como “Ether Dome” ubicado en el Hospital General de Massachusetts, comenzó la búsqueda de entender cómo actúan los anestésicos generales. Tan sólo 6 meses después de la exitosa demostración, los Doctores Emil Harless y Baron Ernst von Bibra al notar que los anestésicos son altamente afines a los tejidos grasos propusieron que los anestésicos extraían los lípidos del cerebro11. No obstante, esta primera explicación al no dar cuenta de como ocurre la reversión del efecto no fue aceptada por la comunidad científica. Estos científicos posteriormente no insistieron en su hipótesis, pero, dos décadas más adelante el Dr. Ludimar Hermann propuso que los anestésicos actuaban como solventes sobre la “lecitina” de las células, la que actualmente conocemos como los fosfolípidos, pero esta propuesta también se desechó rápidamente por los mismos motivos que la primera11. Sin embargo, ambas observaciones tienen el valor de que plantearon a la comunidad científica que los anestésicos tienen una gran afinidad por lo lipídico. Luego, en el contexto de la controversia de finales del siglo XIX en relación a que es lo que se considera vivo, el Dr. Claude Bernard planteó que un ser puede ser considerado vivo si éste tiene la susceptibilidad de ser anestesiado11. Esta conclusión la obtuvo tras observar que diversos seres vivos, tales como humanos, roedores, plantas, perdían la capacidad de reaccionar a los estímulos externos al estar anestesiados o narcotizados como se le denominaba en esa época. Esta hipótesis de Bernard ha promovido la idea de un mecanismo único para explicar la acción de los anestésicos generales, el que debería estar presente en todos los seres vivos. De hecho, el fue pionero al plantear que existen diferentes agentes anestésicos, pero, sólo un estado anestesiado, idea que ha prevalecido hasta nuestros tiempos.
A comienzos del siglo XX, Meyer y Overton realizaron trabajos sistemáticos de manera independiente, en los cuales correlacionaron la potencia anestésica en los renacuajos con la solubilidad de los anestésicos en aceite de oliva11–14. Interesantemente, ambos investigadores encontraron una estrecha correlación positiva entre la potencia y la liposolubilidad de los anestésicos. Aquellos compuestos más liposolubles eran anestésicos más potentes que aquellos menos liposolubles. Esta correlación se conoce como la teoría de Meyer-Overton, sin embargo, algunos autores prefieren denominarla la regla de Meyer-Overton. Estos resultados indican que el mecanismo de acción de los anestésicos generales debiera ser único, como lo había planteado Bernard, y que éste dependía de la liposolubilidad del anestésico. Pese a ello, esta teoría fue abandonada hasta finales de los ‘60, momento en que el Dr. Edmond Eger empezó a establecer como medida de potencia de los anestésicos inhalatorios el MAC (por sus siglas del inglés de minimum alveolar concentration), la que se refiere a la concentración alveolar mínima necesaria para prevenir el movimiento producido como respuesta a un estímulo doloroso15. Interesantemente, al correlacionar el MAC con la liposolubilidad se observó lo mismo que Meyer y Overton habían reportado 70 años atrás. Esto hizo que la teoría de los lípidos retomara relevancia. No obstante a ello, diversos reportes comenzaron a contradecir a la teoría de que los anestésicos actúan sobre los lípidos. En los años ‘70, se demostró que los anestésicos alteraban la luminosidad de las luciérnagas, fenómeno que depende de una proteína denominada luciferesa16. De esta manera, si se aplicaba un anestésico a esta enzima luciferasa se dejaba de producir luz. Relacionada a esta observación, los Doctores Nick Franks y Bill Lieb publicaron el año 1984 un artículo ícono17, el que determinó que la concentración inhibitoria 50 (IC50, por sus siglas del inglés) de diversos anestésicos sobre la luciferesa se correlacionaba estrechamente con su liposolubilidad y con su potencia anestésica. A partir de este estudio la comunidad científica ha aceptado que él o los blancos de los anestésicos generales son proteínas y no lípidos17. Sin embargo, con el transcurso de los años se ha determinado que los anestésicos actúan sobre bolsillos lipofílicos de las proteínas, lo que ha permitido explicar la relevancia de la lipofilicidad en la potencia de los anestésicos y el fenómeno de cutoff observado inicialmente en derivados del alcohol18. Este fenómeno consiste en que ciertos compuestos, como los alcoholes, en la medida que aumenta su liposolubilidad debido al aumento del número de carbonos, la potencia anestésica aumenta hasta cierto punto denominado cutoff. Pasado dicho punto el efecto anestésico se pierde. Esto se explica porque el bolsillo acepta moléculas hasta cierto tamaño, luego por un impedimento estérico, el compuesto con un mayor número de carbonos no lograría entrar en el bolsillo y el efecto se pierde. En resumen, desde la primera demostración exitosa del éter hasta la fecha se ha avanzado en el entendimiento del mecanismo de los anestésicos. Se ha aceptado que estos compuestos actúan sobre bolsillos hidrofóbicos de ciertas proteínas, lo que en términos conceptuales no desecha la teoría de Meyer-Overton, dado que ellos sólo establecieron que la potencia anestésica se correlaciona con la liposolubilidad. Además, hasta el día de hoy la idea de que todos los anestésicos actúan a través de un único mecanismo es un tema de discusión y será tratado en profundidad en la siguiente sección.
TEORÍA UNITARIA Y MÚLTIPLEDesde un inicio se ha planteado que el mecanismo de acción de los anestésicos generales debiera consistir en la acción de estos fármacos sobre un único blanco, el cual debiera dar cuenta de todos los efectos clínicos observados en una AG. Esto se conoce como la teoría unitaria de la acción de los anestésicos generales y se ha mantenido desde las observaciones de Bernard, pasando por la teoría de Meyer-Overton hasta Nick Franks, quien demostró que los anestésicos actúan sobre proteínas. La proteína blanco que ha permitido sostener la teoría unitaria ha sido el receptor γ-ácido amino butírico tipo A (GABAA), dado que la acción de los anestésicos sobre este receptor pareciera ser necesaria y suficiente para inducir una AG19. Sin embargo, en los últimos 20 años, se ha establecido que la acción de los anestésicos generales sobre el receptor GABAA no daría cuenta de todos los efectos clínicos de una AG2,8. Y con estas observaciones, se han establecido las bases de la teoría múltiple, la cual señala que los anestésicos actúan sobre múltiples blancos moleculares para producir los diferentes efectos clínicos.
Diferentes observaciones apoyan a la teoría múltiple de la acción de los anestésicos. Por un lado, se ha demostrado que una misma concentración de un anestésico es capaz de actuar sobre diferentes receptores2,8,9. De hecho, cuando se ha comparado la relación entre la estructura y la actividad de diferentes anestésicos para producir los diferentes efectos clínicos, se han encontrado diferencias que no pueden ser explicadas solamente por diferentes afinidades a un mismo blanco molecular, sino que por la acción sobre diferentes blancos moleculares. Por ejemplo, la relación entre la concentración capaz de producir hipnosis y aquella capaz de producir inmovilidad para el óxido nitroso o el éter es significativamente mayor a la relación que exhiben otros anestésicos volátiles halogenados20,21. Esto sugiere que la hipnosis y la inmovilidad es mediada por blancos moleculares diferentes y no por diferentes afinidades a un mismo blanco molecular. Por otro lado, se ha reportado que la administración de ciertos compuestos que teóricamente debieran comportarse como anestésicos no logran hacerlo debido a que no generan inmovilidad e hipnosis y sólo son capaces de producir amnesia22. A estos fármacos se les ha denominado como compuestos “no-inmovilizadores”23. Este comportamiento farmacológico se podría explicar porque los diferentes efectos clínicos producidos por los anestésicos son el resultado de la acción sobre diferentes blancos moleculares, lo que nuevamente estaría refutando la teoría unitaria. Por último, existen anestésicos generales que no actúan sobre GABAA y son capaces de producir un estado clínico de una AG de manera similar que los fármacos que actúan principalmente sobre los receptores GABAA. Es el caso del xenón, óxido nitroso y de la ketamina, los cuales se saben que son fármacos que actúan sobre los receptores de N-metil-D-aspartato (NMDA) para inducir una AG2,24. En conjunto, estas evidencias apoyan a que los anestésicos actúan sobre múltiples blancos moleculares para dar cuenta de los distintos efectos clínicos y apoyan la visión de la teoría múltiple. No obstante a ello, no es descartable que el estado final que se genera en una AG tenga un único patrón de inhibición de las redes neuronales, el cual podría ser compartido por todos los fármacos independiente de los blancos moleculares. Sin embargo, esta es una pregunta que aún está sin respuesta. A continuación, resumiremos la acción de los principales anestésicos sobre los distintos blancos moleculares conocidos que se han propuesto como los responsables de sus efectos clínicos (Figura 3).
.")
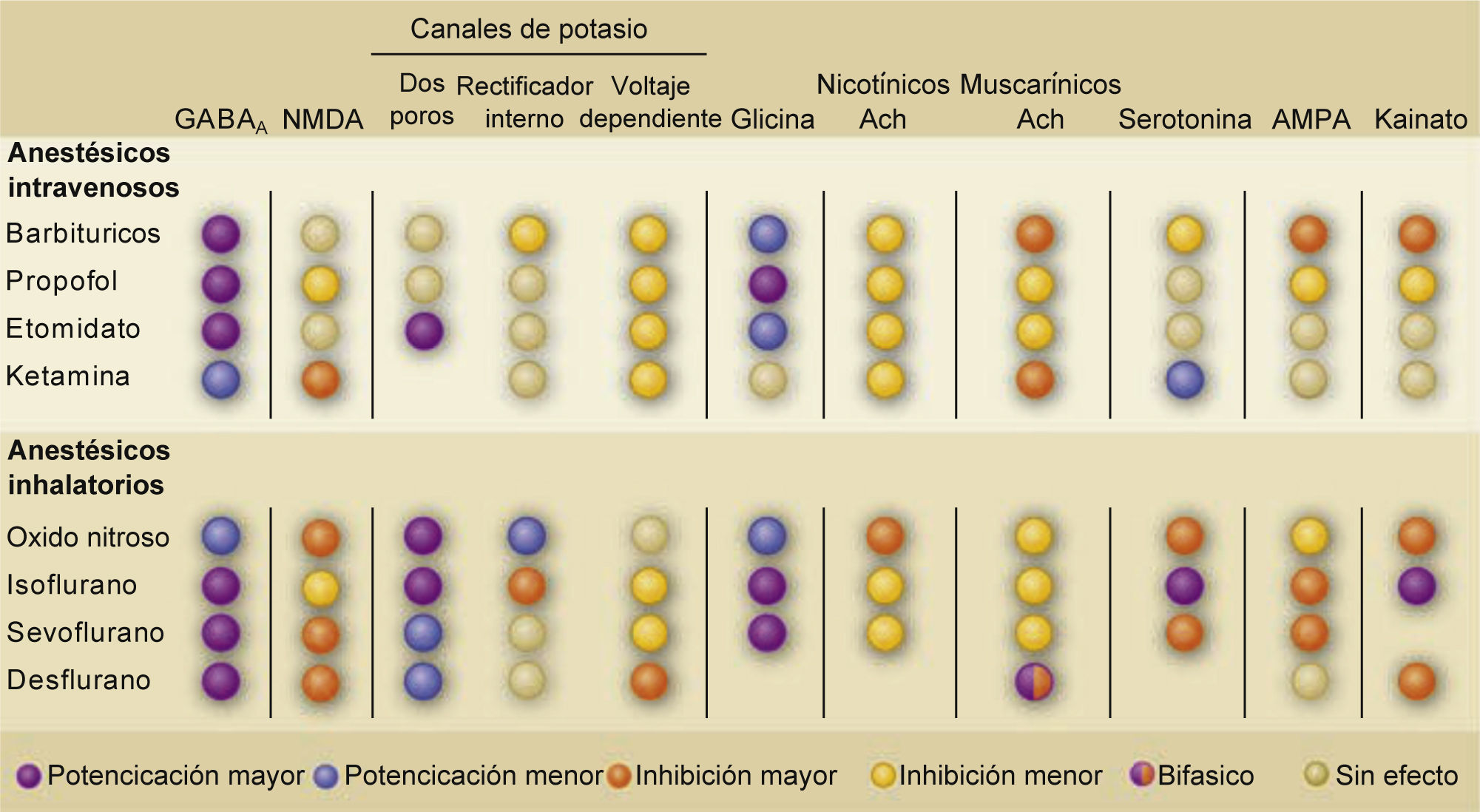
TABLA RESUMEN DE LOS BLANCOS MOLECULARES DE LOS DIFERENTES ANESTÉSICOS GENERALES
Abreviaciones: Ach: acetilcolina; AMPA: ácido α-amino-3-hidroxi-5-metilo-4-isoxazolpropiónico; GABAA: receptores γ-ácido amino butírico tipo A; NMDA: N-metil-D-aspartato. Tomado y modificado de Alkire, M.T., Hudetz, A.G. & Tononi, G. Consciousness and anesthesia. Science 322, 876-880 (2008).
El γ-ácido amino butírico (GABAA) es un aminoácido y constituye el principal neurotransmisor inhibitorio del sistema nervioso central en mamíferos. Se han descrito dos tipos principales de receptores que responden a GABA: los receptores GABAA y los receptores GABAB26,27. Los GABAA corresponden a un receptor de tipo ionotrópico, por lo cual son canales iónicos que al activarse son permeables a cloruro y bicarbonato en una razón de 4:1, o sea, el cloruro es 4 veces más permeable que el bicarbonato26. Por otro lado, los receptores GABAB corresponden a un receptor metabotrópico acoplado a una proteína G inhibitoria25. En esta sección nos referiremos exclusivamente a los receptores GABAA, dado que son el principal blanco molecular de los principales anestésicos, tales como inhalatorios, etomidato, propofol e incluso ketamina27,28.
Los receptores GABAA pertenecen a la superfamilia de receptores Cys-Loop, que se caracterizan funcionalmente por ser canales iónicos activados por ligando y se componen por 5 subunidades organizadas en un complejo heteropentamérico que conforman un poro al centro (Figura 4)26,29,30.

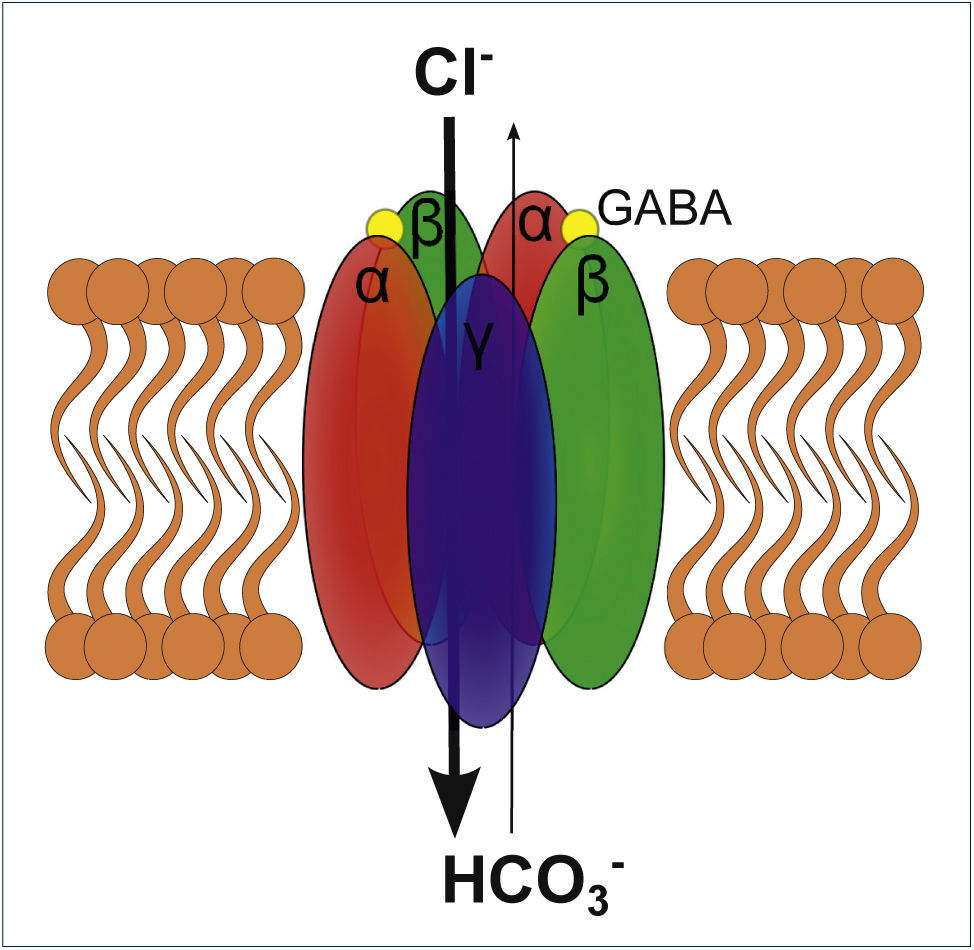
ESQUEMA DE UN RECEPTOR GABAA
El receptor GABAA es un receptor ionotrópico que al activarse permite el paso de Cl− y HCO3, siendo Cl− 4 veces más permeable que HCO3−. El receptor se compone de 5 subunidades transmembrana, las cuales habitualmente son las que se representan en el esquema: αβαβγ. El receptor para activarse requiere de la unión de 2 moléculas GABA, las que se unen en ambas interfaces αβ lo que es representado en el esquema.
A la fecha se han clonado 19 subunidades diferentes para el receptor GABAA (α1-6, β1-3, γ1-3, δ, ¿, π, θ y σ1-3)26–28. Sin embargo, el 60% de los receptores GABAA están compuestos por 2 subunidades α, 2 subunidades β y 1 subunidad γ, en un orden αβαβγ29,30. Cada una de las subunidades se compone de 4 dominios transmembrana, con su amino y carboxilo terminal orientados hacia el extracelular26,30. El ligando endógeno es GABA y se requiere la unión de dos moléculas de GABA en el receptor para que éste se active. Los dos sitios de unión a GABA están localizados en la cara extracelular en cada una de las interfaces entre una subunidad α y una subunidad β26,29,30. Es destacable que las diferentes subunidades le confieren al receptor propiedades biofísicas particulares, y localizaciones determinadas a nivel del sistema nervioso central y a nivel de la sinapsis27,29. Los ligandos exógenos más conocidos de los receptores GABAA son los anestésicos generales, el etanol y las benzodiacepinas. Los anestésicos actúan sobre los receptores GABAA como moduladores alostéricos positivos (PAM, por sus siglas del inglés). Esto quiere decir que los anestésicos requieren GABA endógeno para poder activar al receptor31. No obstante, a muy altas concentraciones se comportan como agonistas directos32. Además, dependiendo las subunidades que componen al receptor GABAA, los anestésicos producirán diferentes niveles de potenciación y con diferentes cinéticas33. Los sitios de unión de los anestésicos en los receptores GABAA se han estudiado con mutaciones sitio dirigidas. De este modo, se ha determinado que la serina 270 de las subunidades α es crítica para el efecto PAM de isofluorano32,34. Por otro lado, la leucina 232 de las subunidades α cuando es mutada se pierde el efecto del halotano34. En cuanto a los anestésicos intravenosos, se sabe que el sitio de acción estaría en las subunidades β2-332,35. Específicamente, el efecto clínico de la administración de propofol o etomidato se pierde al mutar la asparragina 265 de la β235. Posteriormente, con otras mutaciones sitio dirigidas se han ido configurando los bolsillos hidrofóbicos donde actúan los anestésicos generales32.
Los receptores GABAA al activarse disminuyen la capacidad de las neuronas para generar un potencial de acción y, con ello, la transmisión del impulso eléctrico. Esta disminución de la excitabilidad neuronal ocurre porque la activación de GABAA genera una hiperpolarización o una inhibición tipo cortocircuito de la excitabilidad neuronal32,36. Esta inhibición ocurre por la activación de los receptores GABAA sinápticos y extrasinápticos, los primeros son receptores localizados en las sinapsis neuronales y se caracterizan por generar una inhibición tipo fásica y tener una afinidad baja por GABA, mientras los extrasinápticos están localizados alrededor de la sinapsis, generan una inhibición tipo tónica y son altamente afines a GABA27,32,36. Los anestésicos actúan sobre ambos tipos de receptores para generar los diferentes efectos clínicos. Por ejemplo, los receptores que contienen la subunidad α5 son extrasinápticos, se expresan particularmente en las células piramidales del hipocampo, estructura fundamental en la adquisición de la memoria, y explican la amnesia generada por los anestésicos y aún más los posibles déficit de memoria que ocurren transitoriamente tras una anestesia4,37.
Otros blancos molecularesHoy es ampliamente aceptado que los anestésicos no solo actúan sobre los receptores GABAA. En esta sección sólo haremos una breve descripción estructural y funcional de estos blancos moleculares.
Canales de potasio de dos poros: son un subtipo de canales de K+ voltaje dependientes que mantienen el potencial de membrana de reposo por medio de una corriente “background”. Estructuralmente se conforman por 2 subunidades donde cada una contiene 2 dominios de poro, característica que origina el nombre de estos canales. Entonces al ensamblarse estas 2 subunidades, se conforma un único poro permeable a K+ rodeado por 4 dominios de poro, donde cada dominio contiene un sensor de potencial para abrir al canal2,38. Estos canales se expresan en el sistema nervioso central y se han descrito como un blanco de los anestésicos inhalatorios39.
Receptores de N-metil-D-aspartato (NMDA): son receptores ionotrópicos que se activan con glutamato. Estructuralmente son un tetrámero y cada subunidad consta de 4 dominios transmembrana. Desde un punto de vista funcional corresponden a un canal catiónico no selectivo, permeable a cationes mono y divalente, como Na+ y Ca2+. Su apertura lleva al ingreso de cationes a la célula, lo que despolariza a la membrana plasmática y genera un potencial postsináptico excitatorio. Es ampliamente aceptado que constituye un blanco molecular de diversos anestésicos generales, como el xenón, la ketamina y el óxido nitroso2,40,41, los cuales actúan como antagonistas de NMDA inhibiendo la excitación y, con ello, disminuyen la excitabilidad neuronal.
Receptores de glicina: son receptores ionotrópicos pentaméricos, que al igual que los receptores GABAA pertenecen a la familia de los receptores cys-loop. Su ligando endógeno es el aminoácido glicina. Este canal también es permeable a Cl− y su activación genera la inhibición de la excitabilidad neuronal42. De hecho, corresponden al segundo tipo de receptores más importantes en la neurotransmisión inhibitoria del sistema nervioso central. Se expresan preferentemente en el tronco encefálico y en la médula espinal43. Es por esto que se han propuesto como blanco molecular de los anestésicos inhalatorios, los cuales actuarían como PAM sobre estos receptores44, para producir la inmovilidad, pero esto ha sido descartado45.
Canales catiónicos activados por la hiperpolarización y modulados por nucleótidos cíclicos (HCN): son canales expresados en células excitables y se caracterizan por generar actividad de marcapaso. Son activados por la hiperpolarización de la membrana plasmática. Estructuralmente son un tetrámero y cada subunidad consta de 6 dominios transmembrana. Conducen principalmente Na+. La unión de nucleótidos cíclicos como la adenosina monofosfato cíclico (AMPc) alteran la sensibilidad al voltaje del canal, abriéndose a potenciales más despolarizados46. Se ha reportado que los anestésicos inhalatorios pueden modular estos canales dependiendo de las subunidades constituyentes47.
Canales de sodio sensibles a voltaje (Nav): son canales iónicos voltaje dependientes permeables a Na+. Algunos subtipos se expresan en el sistema nervioso central y propagan el potencial de acción. Se ha descrito recientemente que los anestésicos pueden inhibir las corrientes de los canales de Na+ activados por voltaje48,49. Dado que tienen un rol fundamental en la propagación del potencial de acción y en definitiva en determinar la liberación de neurotransmisores, son un blanco atractivo en el estudio del mecanismo de los anestésicos generales particularmente en cómo establecen la inmovilidad los anestésicos inhalatorios50.
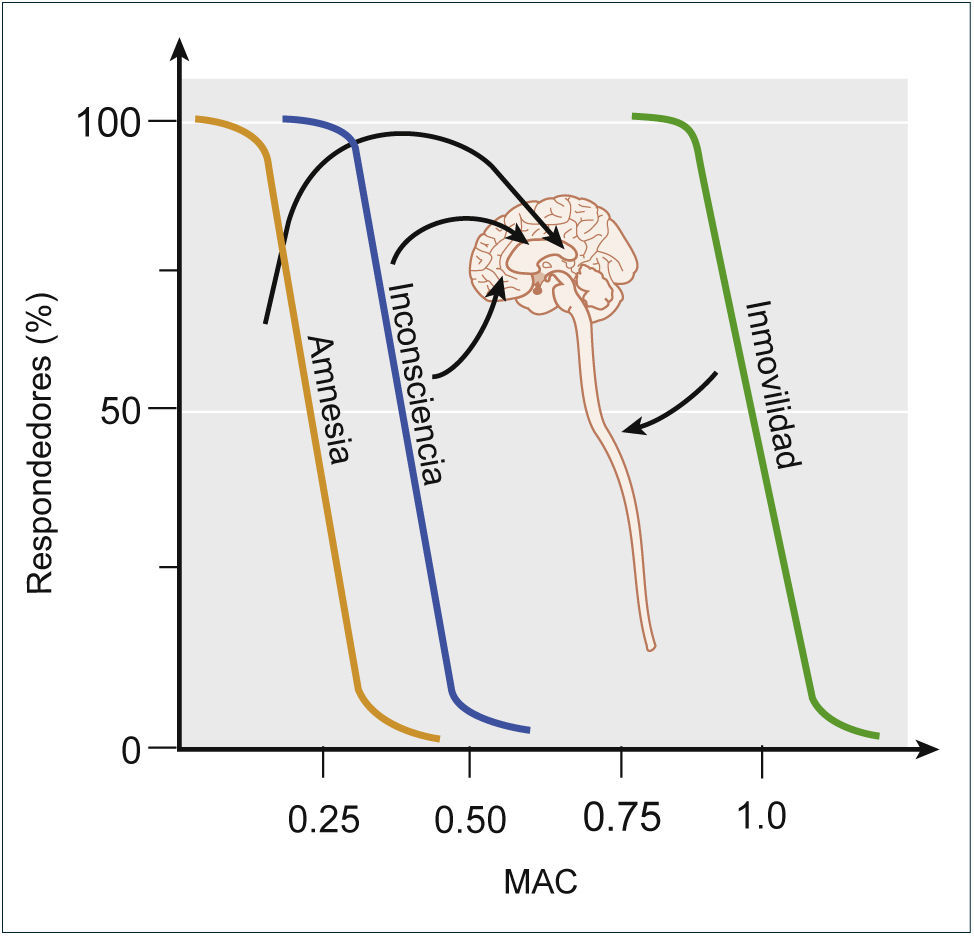
EFECTOS CLÍNICOSComo fue mencionado previamente, los anestésicos generan un estado que se puede definir como un coma farmacológico reversible con una conservación de la homeostasia interna. Este estado se caracteriza por la hipnosis o inconsciencia, la amnesia y la inmovilidad. Estos efectos clínicos reflejan la acción de los anestésicos sobre diversos blancos moleculares, los cuales que se expresan en diferentes estructuras a nivel del sistema nervioso central. Como fue mencionado previamente, la amnesia es el primer efecto que se establece a bajas dosis y/o concentraciones de los anestésicos y el hipocampo es la estructura que la subyace; luego, la inconsciencia se establece a una mayor dosis y/o concentración cuando los anestésicos interactúan con la corteza y estructuras subcorticales; y finalmente, se establece la inmovilidad a altas dosis y/o concentraciones y su sustrato anatómico es la médula espinal (Figura 5)10.

EFECTOS CLÍNICOS Y SITIOS DE ACCIÓN QUE SUBYACEN EL EFECTO DE LOS ANESTÉSICOS INHALATORIOS
La amnesia es el efecto más sensible y es secundaria al efecto de los anestésicos sobre el hipocampo, amígdala, entre otras estructuras. La inconsciencia requiere una mayor dosis y se obtiene por la acción de los anestésicos sobre la corteza, tálamo y estructuras subcorticales. La inmovilidad ocurre por la acción de los anestésicos sobre la médula espinal y requiere una dosis significativamente mayor en relación a los otros efectos. Tomado y modificado de Miller's Anesthesia, Eigth Edition, (2015).
Actualmente no existe un consenso para explicar cómo los anestésicos establecen la inconsciencia, lo que ha motivado el planteamiento de diferentes mecanismos, entre los cuales destacan:
- a)
Los anestésicos actúan a nivel de los núcleos subcorticales que están involucrados en el sueño2,51.
- b)
Los anestésicos generan una desintegración de la interconexión de la corteza cerebral con lo que se hace incapaz de responder a estímulos externos24,52,53.
- c)
Los anestésicos establecen el estado de inconsciencia afectando la corteza y los núcleos subcorticales dependiendo sobre qué blancos moleculares actúan3,54.
La primera propuesta señala que los anestésicos, específicamente los gabaérgicos, actúan a nivel de los núcleos que promueven y mantienen el sueño, particularmente sobre el núcleo preóptico ventrolateral (VLPO, por sus siglas del inglés). Este núcleo libera GABA desde sus terminales localizados en los núcleos que promueven el despertar, núcleo locus ceruleus (LC) y núcleo tuberomamilar (TMN). De este modo, si la actividad del VLPO se hace predominante, entonces se produce la inhibición del LC y TMN estableciéndose el estado de sueño. Se propone que los anestésicos potenciarían la inhibición del VLPO sobre el LC y TMN con lo que se produce el estado de inconsciencia observado en una anestesia51. La segunda hipótesis señala que los anestésicos, independientemente si son gabaérgicos o si actúan sobre los receptores NMDA, desintegran la información en la corteza cerebral, particularmente el flujo de la información que va desde la corteza frontal y prefrontal hacia la corteza parietal y los niveles subcorticales y periféricos, con lo que se establece el estado de no respuesta observado en una AG52. Finalmente, se ha propuesto que cada uno de los anestésicos establece un estado de inconsciencia particular, dependiendo de los blancos moleculares sobre los cuales actúa el anestésico54. Por ejemplo, propofol y etomidato actuarían a nivel cortical promoviendo la inhibición gabaérgica de las interneuronas sobre las neuronas piramidales, y al mismo tiempo potenciaría la actividad GABA en el tálamo y en los terminales de las neuronas del VLPO54,55. De hecho, en estados inflamatorios, en los cuales se promueve un aumento de la expresión de los receptores GABAA en la superficie neuronal, se requiere una menor dosis para generar un estado de AG en ratones56. Por otro lado, debido al antagonismo sobre los receptores NMDA, ketamina establece una inhibición de la actividad glutamatérgica de las interneuronas, con lo cual las neuronas piramidales corticales, neuronas hipocampales y del sistema límbico quedan en un estado excitatorio aberrante incapaz de recibir y responder a los estímulos externos54.
La inconsciencia inducida por los anestésicos en humanos se ha estudiado con el electroencefalograma (EEG), el que da cuenta de la actividad eléctrica cortical. Los cambios observados en el EEG durante una AG han permitido darle un sustento experimental a los mecanismos propuestos de como los anestésicos generan la inconsciencia. En una AG se observa un enlentecimiento de la actividad eléctrica, lo que se refleja en una disminución de la potencia global, la que es cuantificada en el espectrograma obtenido tras el procesamiento de la señal cruda del EEG57. Además, la coherencia, es decir la sincronía, disminuye entre las diferentes zonas en las que se adquiere la señal eléctrica, lo que da sustento a la desintegración cortical generada por los anestésicos58. Además, para cada uno de los fármacos se han determinado diferentes patrones en el espectrograma57. Por ejemplo, los inhalatorios, propofol y etomidato promueven un aumento de la potencia en las bandas de frecuencia α (8-12Hz) y δ (0-4Hz), las que son ondas de mayor amplitud y de menores frecuencias que las ondas observadas en vigilia, ondas de la banda β y γ (13-25Hz y >25Hz, respectivamente)57,59. Interesantemente, tanto con los inhalatorios como con propofol se observa un fenómeno que se ha denominado anteriorización del α, es decir la banda de frecuencia α se presenta a nivel frontal59, lo que no se observa en situaciones fisiológicas. La banda α se presenta habitualmente a nivel occipital al cerrar los ojo. Se presume que esta anteriorización del α estaría dando cuenta del establecimiento de la reverberación del circuito corticotalamico entre la corteza prefrontal y el tálamo60. Es de destacar que esta señal eléctrica disminuye con la edad61,62. Por último, en una AG con agentes antagonistas de los receptores NMDA como ketamina, también se produce un aumento de la señal de baja frecuencia δ, pero además destaca una actividad γ de alta frecuencia, la que subyace la actividad excitatoria aberrante de las neuronas piramidales corticales en ausencia del control de las interneuronas57.
Aunque no exista un consenso de cómo los anestésicos establecen la inconsciencia, sin duda alguna el efecto potenciador o antagonista sobre diferentes blancos moleculares en diferentes redes neuronales corticales y subcorticales es evidente. No obstante, son necesarios más estudios para determinar si existe una red neuronal particular que actúe como un interruptor, de modo que al activarse, independientemente del tipo de anestésico, se establece la inconsciencia.
InmovilizaciónEl efecto inmovilizador se ha estudiado principalmente en los agentes inhalatorios. Esto debido a que el éter y sus derivados establecen la inmovilización a concentraciones que permiten ser utilizados como agentes únicos8,15. Por el contrario, con propofol, como representante de los intravenosos, establece la inmovilización a muy altas concentraciones, 4 veces por sobre la concentración a la que se establece la inconsciencia, por lo que no se utiliza como agente único. Además, se sabe que propofol establece la inmovilización vía potenciación de los receptores GABAA y glicina a nivel medular2,3,32. El mecanismo por el cual los inhalatorios producen la inmovilización aún es parcialmente conocido, pero se sabe que se genera a concentraciones cercanas a la inconsciencia. Se ha establecido que es mediante un mecanismo en la médula espinal y no a nivel cerebral, y que se correlaciona con la disminución de la excitabilidad de las motoneuronas en la médula espinal63,64. Esto sugiere que los inhalatorios son capaces de actuar directamente sobre las motoneuronas. El blanco molecular específico que da cuenta de la inmovilidad ha sido objeto de permanente debate y estudios50, y se han descartado múltiples blancos potenciales y actualmente se desconoce la identidad molecular sobre el que actúan los inhalatorios para generar la inmovilidad. Se han descartado los receptores NMDA de las motoneuronas de la médula espinal, pese a que se propusieron como un posible blanco50. Por otro lado, también se descartó el rol de los receptores GABAA65. Estudios experimentales han mostrado que los receptores de glicina medulares tampoco son el blanco molecular que da cuenta del efecto inmovilizador de los inhalatorios45. Lo mismo ha ocurrido con los receptores nicotínicos de acetilcolina, los receptores de serotonina y los receptores opioides45,50. No obstante a todo lo anterior y pese a que son un blanco poco probable, se especula que los Nav podrían ser el blanco molecular tan elusivo de encontrar48–50, pero se requieren de nuevos estudios.
AmnesiaLa adquisición de memoria depende del hipocampo, la amígdala, la corteza frontal, entorrinal y perirrenal. Las neuronas piramidales del hipocampo expresan receptores GABAA extrasinápticos que contienen la subunidad α5 (α5GABAA), los cuales son críticos en modular la adquisición de la memoria66. De modo que cuando hay un estímulo suficientemente intenso la inhibición tónica mediada por estos receptores GABAA es sobrepasada, con lo que el nuevo estímulo se puede adquirir como memoria66. Por el contrario, si el estímulo es débil esto no ocurre. Los anestésicos potencian a los receptores α5GABA y se ha determinado que son los blancos moleculares que median la amnesia mediada por etomidato37. Preocupantemente, estos receptores α5GABAA tras una dosis única de etomidato o una exposición breve con isofluorano aumentan su expresión en la membrana plasmática, tras lo cual la adquisición de nueva memoria empeora. Este fenómeno se ha observado que es transitorio y es prevenido con antagonistas específicos de estos receptores4.
CONCLUSIONESDesde la primera demostración pública exitosa el año 1846 hasta nuestro días ha habido un gran avance en el conocimiento del mecanismo de los anestésicos. Cada conocimiento nuevo se ha basado en el previo, con lo cual se han ido planteando hipótesis más certeras. No obstante, la idea fundamental planteada por Bernard de que un ser vivo para considerarse como tal debe tener la susceptibilidad de ser anestesiado y que existe un único estado de narcosis pese a que existan múltiples fármacos, nos debe retrotraer a analizar el problema de manera más global y de este modo podremos darle respuestas a estas observaciones fundamentales. Este es un desafío neurocientífico mayor, el cual pude ser abordado con el conocimiento que se tiene actualmente sumado a la práctica diaria en cada uno de los pabellones quirúrgicos.
Cuando un paciente es sometido a una cirugía que requiere una AG se demuestra inequívocamente el mecanismo de acción de los anestésicos. Por ello, los anestesiólogos tienen la capacidad práctica de comprender las funciones cerebrales superiores y sus bases neurocientíficas. Solamente variando la velocidad de una inducción anestésica se puede diferenciar claramente como se establecen los diferentes efectos, amnesia, inconsciencia e inmovilidad y para lograrlos se requieren dosis crecientes, las cuales son similares entre los diferentes pacientes indicándonos que los mecanismo son altamente conservados entre las diferentes personas. Además, pese a que se señala que los diferentes anestésicos alcanzan los mismos efectos clínicos, es evidente en la práctica diaria que los estados de inconsciencia entre los diferentes fármacos, propofol, ketamina, etomidato, inhalatorios, no son iguales. Esto puede considerarse un detalle, sin embargo, da sustento a que los diferentes fármacos actúan sobre diferentes blancos moleculares y sobre diferentes redes neuronales. Del mismo modo, con la incorporación de los monitores de la actividad eléctrica cerebral, lo que sin duda alguna será un estándar de nuestra práctica clínica en un futuro cercano, ha ido permitiendo obtener un conocimiento del efecto de los anestésicos sobre el EEG lo que ha promovido el planteamiento de nuevas preguntas relevantes, no sólo para el cuidado clínico de los pacientes, sino que también para comprender que ocurre con el cerebro bajo los efectos de los anestésicos generales y de este modo dar un paso más en el entendimiento de las funciones cerebrales superiores.
Los autores declaran no tener conflictos de interés, en relación a este artículo.