





Los tumores del sistema nervioso central (SNC) se clasificaron de acuerdo a su histología hasta el año 2007, según la norma de la Organización Mundial de la Salud (OMS). Recientemente se publicó la nueva clasificación de tumores del SNC (2016), con la incorporación de patrones genéticos y moleculares en el diagnóstico de las entidades antes descritas en la versión previa. Dicha modificación con la incorporación de sellos genéticos ha significado una revolución en el diagnóstico de tumores. La nueva clasificación de la OMS incluye modificaciones en gliomas difusos, meduloblastomas y otros tumores embrionarios. Nuevas entidades fueron descritas y algunos conceptos obsoletos sin relevancia clínica fueron removidos. La biología molecular aporta valor en cuanto al pronóstico y el tratamiento de ciertos tumores con comportamientos variados en distintos pacientes (gliomas difusos y glioblastomas). Otro cambio importante fue la incorporación de la invasión cerebral como criterio mayor para el diagnóstico de meningioma atípico grado II. En Chile ya se están implementando estos avances diagnósticos en centros de alta complejidad del país, entre ellos Clínica Las Condes.
Tumors of the central nervous system (CNS) have been classified according to their histologic features as the standard of the World Health Organization (WHO) until 2007. Recently a new classification of tumors of the CNS (2016) was published, adding genetic and molecular features to entities described in the previous version. The incorporation of diagnostic molecular signatures has meant a revolution in the diagnosis of CNS tumors. The new WHO classification includes modifications to diffuse gliomas, medulloblastomas and other embryonal tumors. New entities were described and others were removed, specially those without clinical relevance. Molecular biology provides value in prognosis and treatment of certain tumors with uncertain behavior in different patients, such as diffuse gliomas and glioblastomas. Another important change was the addition of brain invasion as a major criteria for the diagnosis of atypical meningioma grade II. In Chile there are already a few health centers working in the development of these diagnostic advances, including Clinica Las Condes.
Las neoplasias del sistema nervioso central (SNC) constituyen menos del 2% de la totalidad de los tumores en el ser humano. Sin embargo, suelen afectar a niños y personas en edad productiva, a veces con consecuencias devastadoras 1.
Desde su origen en 1979, la organización mundial de la salud (OMS) ha editado 4 textos conocidos como el “libro azul” para clasificar los tumores del SNC 2. Hasta el año 2014, estos tumores se diagnosticaban sólo con técnicas histopatológicas de rutina (tinción de hematoxilina/eosina) e inmunohistoquímica, conservando así cierto criterio de subjetividad del observador. En el año 2016 la OMS publica la nueva clasificación de los tumores del SNC, incorporando estudios de biología molecular en el diagnóstico anatomo-patológico de los tumores primarios del SNC, algo impensado tan solo años atrás 3.
Historia de los tumores glialesLos gliomas son los tumores primarios más frecuentes del SNC y forman un grupo heterogéneo de neoplasias con múltiples tipos histológicos y grados de malignidad 4. Se originan a partir de células progenitoras de la glía, cuyo desarrollo asemeja en la mayoría de los casos una estirpe astrocitaria u oligodendroglial 5. Dichos tumores pueden infiltrar difusamente el parénquima cerebral o situarse solo focalmente, habiendo una correlación genética con su comportamiento biológico en cada escenario 3.
Desde 1884, año en que se diagnosticó y trató quirúrgicamente el primer glioma, estas neoplasias han demostrado un pronóstico pobre, con un crecimiento agresivo y una sobrevida que en el mejor de los casos alcanza los 2 años para lesiones de grado mayor 6.
Las limitaciones de la neurocirugía de inicios del siglo XX hacían que el tratamiento quirúrgico de estos tumores en ciertos casos fuese peor que la evolución natural de la enfermedad. Sin embargo, era éste el único tratamiento posible, incluyendo extensas lobectomías e incluso hemisferectomías, las que además de generar graves secuelas motoras no aportaban mayor sobrevida 6.
Con el advenimiento de nuevas tecnologías, los escenarios quirúrgicos han cambiado, logrando procedimientos cada vez más efectivos y con mejores resultados funcionales postoperatorios. Entre estos cambios se incluyen la monitorización córtico-subcortical, el uso de anestésicos más seguros, la cirugía vigil (tumores ubicados cerca del área del lenguaje) y cuidados post-operatorios en modernas unidades de cuidados intensivos. Además los avances en el entendimiento biológico de estas neoplasias han generado nuevas herramientas terapéuticas, con la creación del término politerapia, que incluye cirugía, radioterapia y quimioterapia 6.
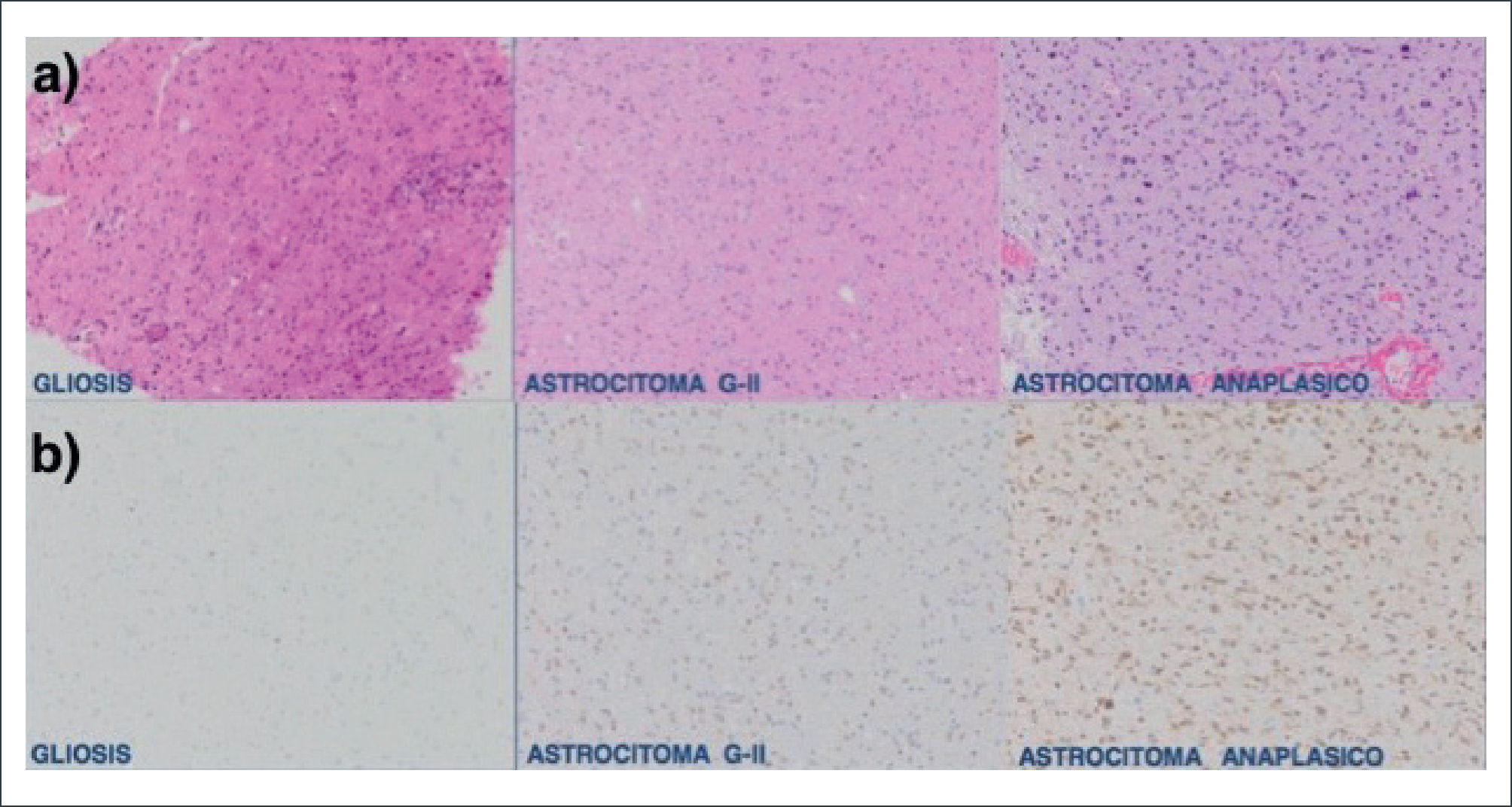
Biología Molecular en GliomasQuizás uno de los descubrimientos más importantes en la biología molecular de los gliomas se produce en el año 2010 con la identificación de mutaciones en el gen de la isocitrato deshidrogenasa IDH 1 y 2, ubicados en el codón 132 y 172 respectivamente. La mutación de este gen es positiva en la gran mayoría de los gliomas infiltrativos difusos (astrocitomas y oligodendrogliomas) así como en un 10% de los glioblastomas 7,8. Múltiples estudios relacionan la presencia de dicha mutación con un comportamiento menos agresivo de estos tumores, diferenciándolos además de otras lesiones con similar histopatología 9–11. Un ejemplo de este hecho es la diferenciación entre gliosis, que es la reacción de las células de la glía ante cualquier noxa al tejido nervioso, versus un astrocitoma difuso grado II 12. En la figura 1 se muestra la expresión de la mutación IDH en un astrocitoma y la ausencia de expresión en gliosis inespecífica 11,12.
 Morfología con técnica convencional de hematoxilina eosina. b) Técnica de IDH-1 mostrando ausencia de tinción en gliosis reactiva y presencia en lesiones de tipo Astrocitoma difuso y Astrocitoma anaplasico.")
Otro problema frecuente para el neuropatólogo avezado es definir si un glioma infiltrativo se asemeja a la estirpe astrocitaria u oligodendroglial. Gracias al aporte de la biología molecular, se ha identificado co-deleción de los brazos de los cromosomas 1p y 19q en neoplasias oligodendrogliales, con implicancias terapéuticas y pronósticas. Por otra parte, la ausencia de la co-deleción favorece el diagnóstico de astrocitoma 14. Además, la presencia de la co-deleción 1p 19q tiene implicancias clínicas. Trabajos prospectivos randomizados demostraron mejor respuesta en el tratamiento con poliquimioterapia conocida como PCV (procarbazina/lomustina/vincristina) versus el uso de radioterapia únicamente, con una media de seguimiento de 10 años 13,14.
En pacientes con glioblastomas, avances tecnológicos en terapias “target” han mejorado la sobrevida en algunos casos. La presencia de metilación del gen MGMT se ha asociado a mayores beneficios en la terapia específica con el uso de Temozolamida, versus aquellos tumores con ausencia de la metilación, demostrado en estudios prospectivos randomizados en concomitancia al uso de radioterapia post-operatoria 15,16.
Importancia del problemaLos tumores del SNC representan aproximadamente un 1.4% de todas las neoplasias, causando un 2.4% de muertes por cáncer al año en los EE.UU. 1. Los casos nuevos en EE.UU. en el año 2016 son 23770, con un número estimado de muertes de 16050 muertes/año 1.
En Chile datos aportados por el proyecto GLOBOCAN de la Agencia Internacional de Investigación sobre el Cáncer (International Agency for Research on Cancer, IARC) refirió los siguientes datos para el año 2012:
Incidencia: 4.6/100000 habitantes. Promedio 2.2% de todos los tumores. Total: 886 casos nuevos/año.
Mortalidad: 2.4/100000muertes. Promedio: 2.0% de las muertes por cáncer. Total: 496 muertes/año.
Prevalencia a 5 años: 9.2/100.000 hab-5 años. Promedio: 1.4%. Total: 1263 casos/5 años 9. La tasa mundial de incidencia según el proyecto GLOBOCAN es de 256213 casos nuevos/año y la mortalidad global 189382 muertes/año 17.
Los tumores primarios del encéfalo más frecuentes son gliomas difusos (astrocitomas y oligodendrogliomas), glioblastomas, gliomas de bajo grado, linfomas del SNC, meningiomas y adenomas hipofisiarios. En la médula espinal los más frecuentes son los schwannomas, ependimomas y meningiomas (constituyendo el 80% de los tumores de la medula espinal). Gliomas, cordomas y tumores vasculares son más inusuales 18.
Las metástasis abarcan aproximadamente un 50% de los tumores del encéfalo, siendo mucho menor los casos en médula espinal 19.
En Chile las garantías explícitas en salud (GES) incorporan el diagnóstico y tratamiento de tan solo cuatro tumores primarios: meningiomas, adenomas hipofisiarios, craneofaringiomas y hemangioblastomas 20. Esto significa un problema para todos aquellos pacientes con tumores fuera del grupo GES. En otras palabras, más de un 50% del total de los tumores del SNC 21 no cuentan con apoyo GES en el diagnóstico molecular, imposibilitando el uso de terapias target que han mostrado mejorar la sobrevida de estos pacientes 12,14–16.
PRINCIPALES MODIFICACIONES EN LA CLASIFICACIÓN DE TUMORES DEL SNCComo se mencionó anteriormente, el diagnóstico de neoplasias del SNC con la antigua clasificación de la OMS del año 2007 se basaba principalmente en microscopía óptica, tinción de rutina con hematoxilina/eosina, técnicas inmunohistoquímicas y las características ultraestructurales apreciadas en la microscopía electrónica en casos aislados 3.
En el año 2014, el consejo de Harlem (Holanda) reunió a más de 20 países y 117 colaboradores a cargo por la Sociedad Internacional de Neuropatología (International Society of Neuropathology). Dicho grupo de expertos comenzó a trabajar en la nueva clasificación de la OMS, con el fin de incorporar avances en la biología molecular de los tumores del SNC. Muchos de estos cambios fueron producto de extensas investigaciones genéticas acerca del origen de dichas neoplasias, su comportamiento según el patrón molecular que esta exprese y la viabilidad terapéutica al atacar ciertas mutaciones con nuevas quimioterapias como el uso de Temozolamida en los gliomas difusos y glioblastomas 3,16.
A continuación mencionaremos algunas de las más importantes modificaciones consensuadas por el consejo de Harlem en el diagnóstico de tumores del SNC.
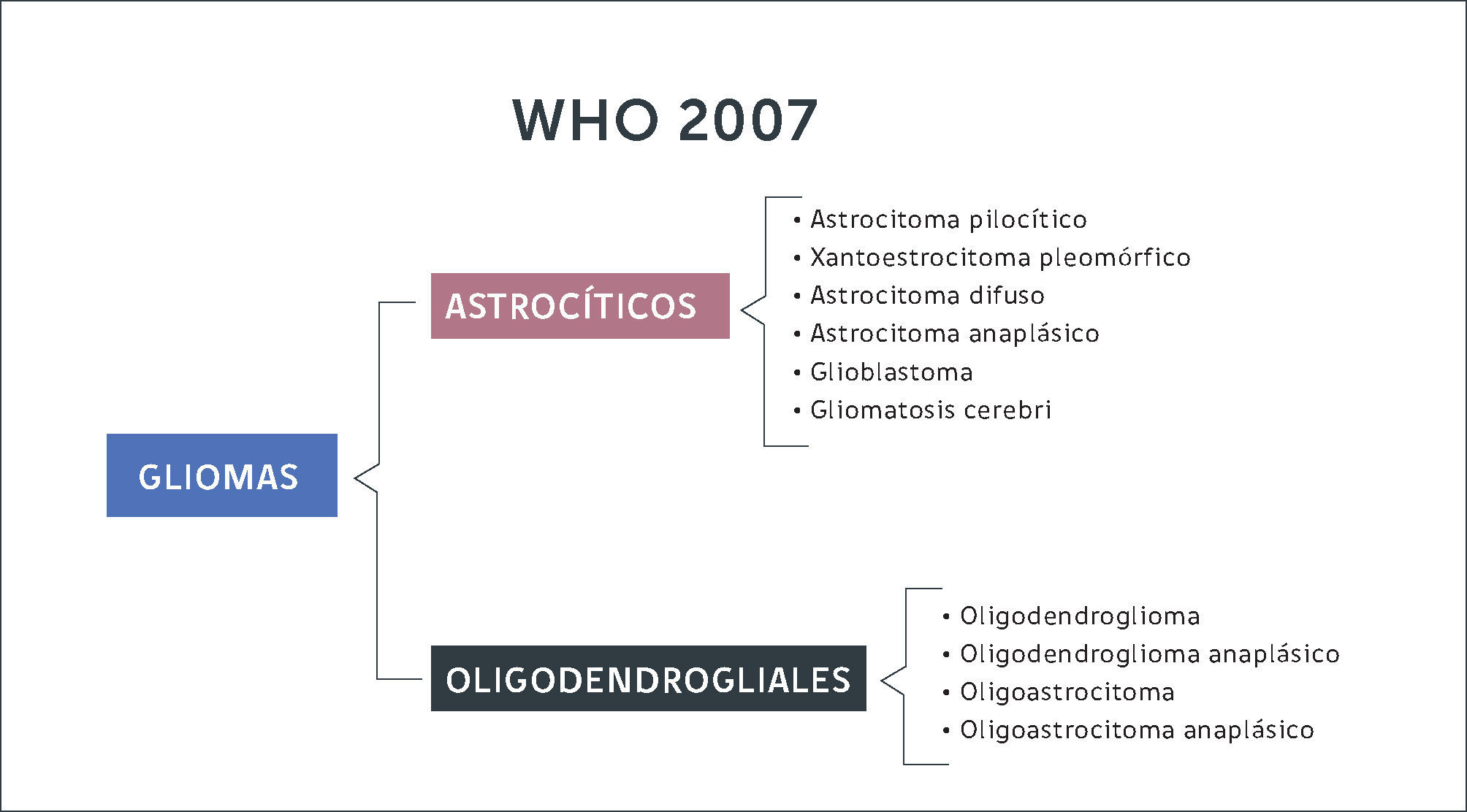
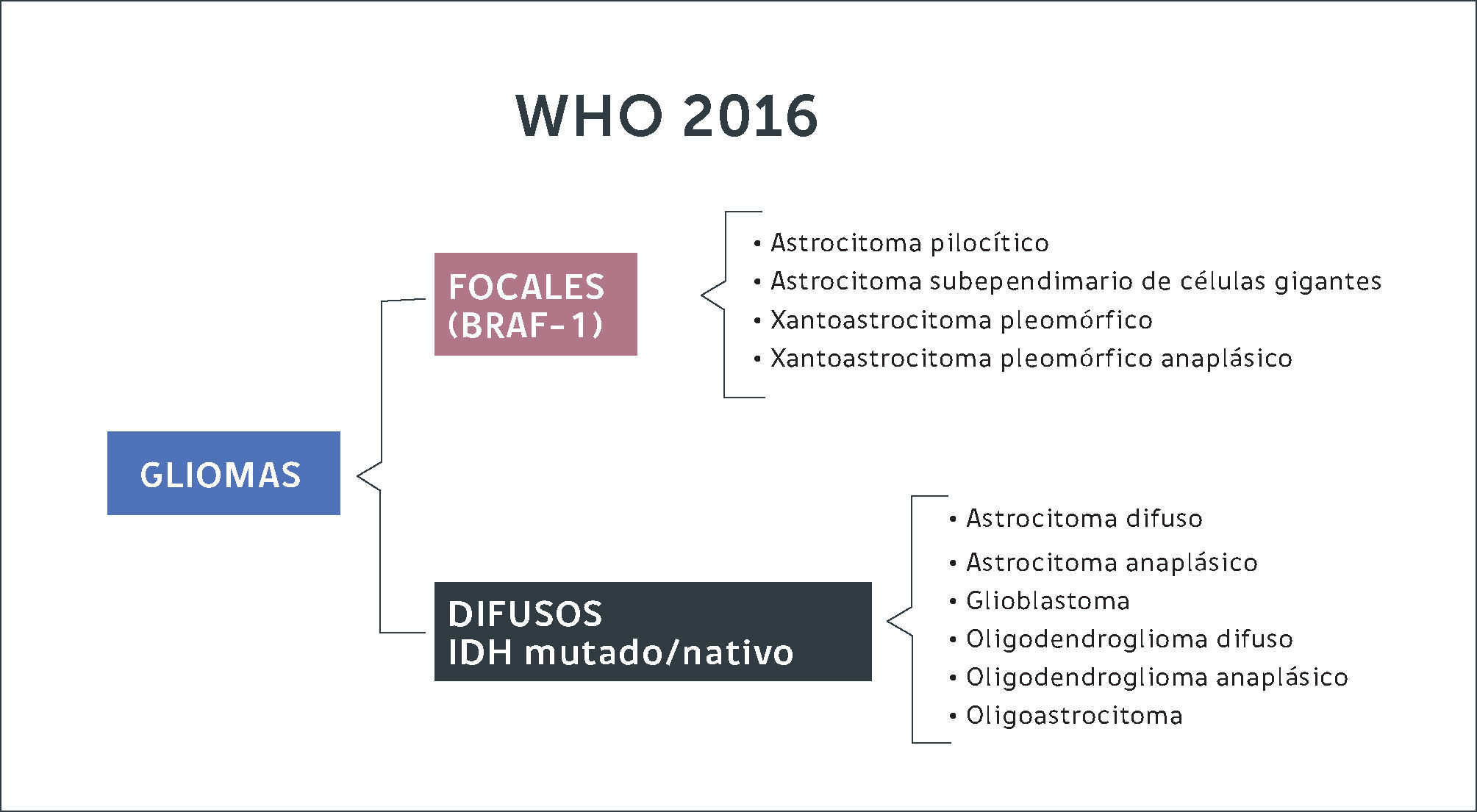
Gliomas DifusosLas células gliales que dan origen a estos tumores pueden originarse a partir de astrocitos u oligodendrocitos. Antiguamente la clasificación del 2007 dividía estos tumores según morfología de la estirpe celular (Figura 2). Con la inclusión de la biología molecular, se logran diferenciar estas entidades según su expresión genética. En resumen, de acuerdo a la nueva clasificación los gliomas se diferencian en gliomas focales de bajo grado (astrocitomas pilociticos, xantoastrocitomas pleomorficos) cuando expresan mutaciones en el gen BRAF-1, estando el gen IDH en genotipo nativo (no mutado). Por otra parte, se constituye el grupo de gliomas de infiltración difusa (astrocitomas, oligodendrogliomas, oligoastrocitomas) subdividiéndolos según estirpe celular y la expresión de la mutación de IDH y co-delecion 1p 19q (Figura 3). En otras palabras, la nueva clasificación de tumores del SNC de la OMS (2016) separa estos tumores según la expresión de estos genes, ya que se correlacionan con pronósticos diferentes.
 Consideraba a los gliomas según estirpe celular en astrocitarios u oligodendrogliales.")
 expresan BRAF-1. En cambio los difusos (grados II a IV) revelan mutaciones en IDH-1 y/o codeleción 1p19q en el caso de los oligodendrogliomas.")
La importancia de la mutación del gen IDH cobra gran relevancia pronóstica según varios estudios. Por ejemplo, la diferencia pronóstica entre el astrocitoma difuso grado II versus anaplásico grado III no es tan significativa si ambos tienen status IDH-mutado 22,23. En ambos casos la presencia de dicha mutación otorga un pronóstico más favorable.
Otro cambio de la nueva clasificación es la eliminación del término Gliomatosis cerebri como entidad diagnóstica aparte. Actualmente se reserva dicho término para un patrón estructural de los gliomas difusos cuando afectan tres o más lóbulos cerebrales, frecuentemente bilateral con crecimiento y regular extensión a núcleos basales y estructuras infratentoriales 3.
GlioblastomaComo se mencionó anteriormente, es sabido desde el siglo XIX el mal pronóstico de esta neoplasia. Estudios en biología molecular han logrado dividir este tumor en dos grandes grupos: IDH nativo (no mutado) versus IDH-mutado. La mayoría de los casos constituyen formas no mutadas o nativas (90% aproximadamente), correspondiendo al tumor de novo o glioblastoma primario, más frecuente en población sobre los 55 años, cuya sobrevida no supera los 15 meses promedio con tratamiento completo (Cirugía + Radioterapia + Quimioterapia) 16,24.
La forma mutada, también conocida como Glioblastoma secundario, es responsable de un 10% de los casos, presentándose en pacientes más jóvenes. Se postula que el origen de este tumor corresponde a la evolución natural (¿Desdiferenciación?) un tumor de menor grado, de ahí el término secundario. En estos casos la presencia de mutación del gen IDH se asocia a una sobrevida mayor (más de 2 años y medio con tratamiento completo) 24–27. En la tabla 1 se encuentra un resumen de las características de ambos tipos.
GLIOBLASTOMAS IDH-NATIVO VERSUS GLIOBLASTOMAS IDH-MUTADO
| GlioblastomaIDH-nativo | GlioblastomaIDH-mutante | |
|---|---|---|
| Sinónimo | Glioblastoma primario | Glioblastoma secundario |
| Precursos | De novo | Astrocitoma difuso Astrocitoma anaplásico |
| Proporción de GB | ∼90% | ∼10% |
| Promedio de edad | 62 años | 44 años |
| Proporción hombre/mujer | 1.42:1 | 1.05:1 |
| Tiempo de síntomas previo a diagnóstico | 4 meses | 15 meses |
| Sobrevida (cirugía + RDT) | 9.9 meses | 24 meses |
| Sobrevida (cirugía + RDT + QMT) | 15 meses | 31 meses |
| Necrosis | Extensa | Limitada |
| Localización | Supretentorial | Preferentemente frontal |
Los meningiomas son tumores de las células meningoteliales de la capa aracnoidal que agrupan aproximadamente un tercio de los tumores primarios del SNC 3. Están incluidos en el programa nacional AUGE 20, tanto su diagnóstico como tratamiento (cirugía y radioterapia complementarias en algunos casos) 28–30.
En cuanto a la nueva clasificación de la OMS, la única variación es la incorporación de la presencia de invasión cerebral como criterio mayor para el diagnóstico de meningioma atípico 3. En la tabla 2 se mencionan los criterios mayores y menores de meningiomas grado II de la OMS (2016).
CRITERIOS MENINGIOMA ATÍPICO (OMS 2016)
| Criterios mayores | Criterios menores |
|---|---|
| • 4 o más mitosis (en 10 campos de aumento mayor) • Invasión cerebral | • Necrosis • Nucléolo prominente • Alta celularidad • Células pequeñas con núcleo prominente (Aumento de la relación núcleo/citoplasma) • Pérdida del patrón estructural |
Para ser considerados atípicos basta con tener 1 criterio mayor o 3 criterios menores.
Los ependimomas son tumores originados a partir del epitelio ependimario. En general son de lento crecimiento, de ubicación infratentorial en relación al 4° ventrículo (niños) o en relación al canal ependimario de la médula espinal (adultos jóvenes) 31.
Desde el punto de vista histológico en ocasiones es complejo diferenciar entre los ependimomas grado II o grado III (anaplásico), estando en discusión el valor clínico de dicha separación. Estudios genéticos se están llevando a cabo para entender la compleja naturaleza biológica de estos tumores 32.
En un estudio de Parker et al se logró identificar un subtipo de ependimomas con fusión del gen RELA. Esta variante correspondería al tumor supratentorial más frecuente en niños, abarcando dos tercios de los ependimomas supratentoriales. La expresión de este oncogen podría eventualmente ser un objetivo en terapia target de este tipo de tumores. Estudios moleculares se están llevando a cabo para dicho tratamiento 33.
MeduloblastomaEs el tumor embrionario del SNC maligno más frecuente del encéfalo pediátrico. Puede tener metástasis “en gota”, es decir, a través del líquido cefalorraquídeo en el resto del neuroeje. Una complicación frecuente es la hidrocefalia obstructiva por ocupación del IV ventrículo 31. Histológicamente se definen 3 grupos con utilidad clínica: clásico, desmoplásico/nodular y de células grandes/anaplásico. El estudio molecular de estos tumores ha permitido clasificarlos en 4 grupos: WNT-activado; SHH-activado, Grupo 3, Grupo 4 34.
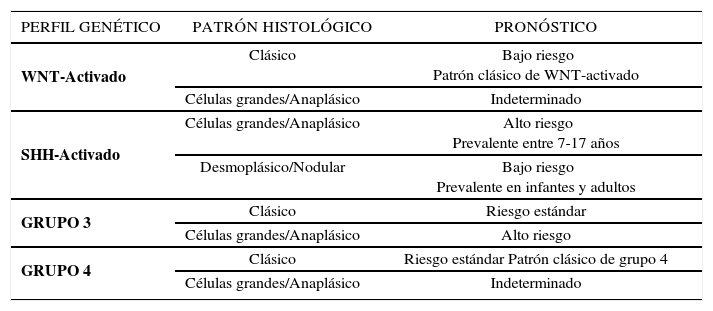
La combinación entre los grupos moleculares con los patrones histológicos ha permitido una aproximación en cuanto a pronóstico y tratamiento específico de estos tumores, siendo los de peor pronóstico aquellos del grupo molecular 3 con patrón de células grandes/anaplásico y el grupo SHH-activado con patrón de células grandes/anaplásico. En la tabla 3 se resumen las combinaciones y pronósticos aproximados.
MÉDULOBLASTOMAS
| PERFIL GENÉTICO | PATRÓN HISTOLÓGICO | PRONÓSTICO |
|---|---|---|
| WNT-Activado | Clásico | Bajo riesgo Patrón clásico de WNT-activado |
| Células grandes/Anaplásico | Indeterminado | |
| SHH-Activado | Células grandes/Anaplásico | Alto riesgo Prevalente entre 7-17 años |
| Desmoplásico/Nodular | Bajo riesgo Prevalente en infantes y adultos | |
| GRUPO 3 | Clásico | Riesgo estándar |
| Células grandes/Anaplásico | Alto riesgo | |
| GRUPO 4 | Clásico | Riesgo estándar Patrón clásico de grupo 4 |
| Células grandes/Anaplásico | Indeterminado |
Los avances contemporáneos en genética y biología molecular se han traducido en un renacimiento en el estudio de los tumores del SNC. Estos nuevos enfoques sugieren el uso de una taxonomía molecular que promete influenciar el diagnóstico, la clasificación de la enfermedad y el manejo clínico. Más aún, el conocimiento y la identificación de sellos moleculares de cada grupo de neoplasias del SNC puede conducir a la expansión del arsenal terapéutico disponible para tratar estas lesiones, brindando así nueva luz de esperanza en el entendimiento y pronóstico de este complejo grupo de lesiones.
Los autores declaran no tener conflictos de interés, en relación a este artículo.