



Los avances en el estudio genético de las enfermedades neuromusculares hereditarias (ENM), a través de variadas metodologías de biología molecular, han permitido arribar, en un número creciente de pacientes, a un diagnóstico etiológico de certeza, acortar el tiempo de estudio en cada caso, ofrecer un correcto asesoramiento genético familiar, establecer relaciones genotipo-fenotipo precisas que brinden eventualmente pautas de pronóstico y tratamiento personalizados, agrupando además cohortes de pacientes para su potencial participación en protocolos clínicos específicos y/o para recibir tratamientos farmacológicos personalizados. En este artículo presentamos un resumen de los estudios de biología molecular usados en pacientes afectados de las ENM más frecuentes. El artículo está fundamentalmente dirigido a neurólogos clínicos, ya sean estos pediátricos o de adultos, quienes, frente a un paciente en el que sospechan una ENM, deben solicitar al laboratorio de biología molecular el estudio genético más apropiado, analizar con el paciente el resultado obtenido y complementar la conducta terapéutica basándose en dicho resultado. El estudio genético solicitado debe brindar las mejores posibilidades diagnósticas para el paciente, en el menor tiempo y con el menor costo posible, beneficiando así al paciente y a los sistemas de salud.
Advances of modern molecular biology methodologies for the genetic study of patients with hereditary neuromuscular diseases (NMD), has resulted in a number of benefits for an increasing number of patients: accurate etiological diagnosis, shorter diagnosis times, proper genetic counseling, precise genotype-phenotype relationships that allow for adequate prognosis and treatment options, and the possibility to group cohorts of patients for their potential participation in specific clinical protocols and/or to receive personalized pharmacological treatments. In this article, we present a summary of the molecular biology studies used in patients affected by the most frequent forms of NMD. Our aim is to assist pediatric or adult neurologists to request the most appropriate genetic study when treating a patient with a potential NMD, and to critically analyze the corresponding results to offer appropriate therapeutic alternatives. The requested genetic study must provide the best diagnostic possibilities for the patient, in the shortest time and at the lowest possible cost, thus benefiting the patient and the health systems.
Los marcados avances en las estrategias de estudios de biología molecular se remontan a la invención de dos metodologías claves, diseñadas por F. Sanger y K.B. Mullis, en los años 1977 y 1985, respectivamente, las cuales permitieron secuenciar de manera relativamente sencilla las moléculas de ADN, así como amplificarlas a través de la reacción en cadena de la polimerasa (PCR). Avances posteriores llevaron a un megaproyecto de colaboración internacional, con financiamiento de sectores públicos y privados por un costo de 3000 millones de dólares, que permitió obtener la primera secuencia completa del genoma humano1,2. El relativamente reciente desarrollo de metodologías de secuenciación masiva de ADN (llamadas de NGS o Next Generation Sequencing), así como los importantes desarrollos en el análisis bioinformático de datos, han permitido estudiar múltiples genes simultáneamente, algunos de gran tamaño y/o extrema complejidad3.
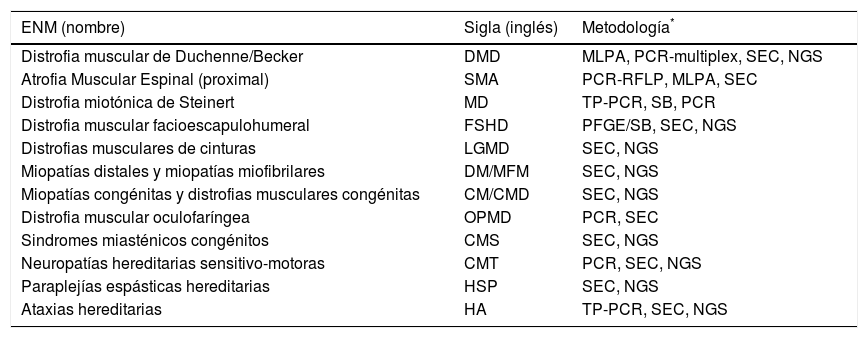
Las enfermedades neuromusculares hereditarias (ENM) constituyen un grupo complejo de entidades clínicas con lesión primaria del músculo y/o del sistema nervioso central y/o periférico (http://www.musclegenetable.fr/) y cuyo diagnóstico definitivo es genético, por lo cual es realizado a través de análisis de biología molecular (Tabla 1). Las ENM pueden tener inicio en el período neonatal, la infancia, la juventud o la vida adulta y mostrar un patrón de herencia autosómico dominante (AD), autosómico recesivo (AR), ligado al cromosoma X o de herencia mitocondrial. La complejidad clínica y genética de las diferentes ENM requiere, para su estudio, de un abordaje multidisciplinario del equipo médico, incluyendo una estrecha interacción entre el neurólogo y el profesional del laboratorio de biología molecular4. Algunas ENM con presentación clínica similar son causadas por mutaciones en genes diferentes, fenómeno conocido como de heterogeneidad genética; por otra parte, mutaciones en un mismo gen pueden dar lugar a ENM con presentaciones clínicas diferentes, fenómeno conocido como de heterogeneidad clínica5. El sostenido estudio de pacientes y familias con ENM, ha conducido a la identificación de 492 genes, para un total de 884 ENM (http://www.musclegenetable.fr/). Alteraciones en genes que codifican proteínas de la matriz del músculo, del sarcolema o la membrana nuclear, del sarcómero, la vaina de mielina, u otras, constituyen la causa de las distintas formas de ENM.
Enm y metodologías diagnósticas
| ENM (nombre) | Sigla (inglés) | Metodología* |
|---|---|---|
| Distrofia muscular de Duchenne/Becker | DMD | MLPA, PCR-multiplex, SEC, NGS |
| Atrofia Muscular Espinal (proximal) | SMA | PCR-RFLP, MLPA, SEC |
| Distrofia miotónica de Steinert | MD | TP-PCR, SB, PCR |
| Distrofia muscular facioescapulohumeral | FSHD | PFGE/SB, SEC, NGS |
| Distrofias musculares de cinturas | LGMD | SEC, NGS |
| Miopatías distales y miopatías miofibrilares | DM/MFM | SEC, NGS |
| Miopatías congénitas y distrofias musculares congénitas | CM/CMD | SEC, NGS |
| Distrofia muscular oculofaríngea | OPMD | PCR, SEC |
| Sindromes miasténicos congénitos | CMS | SEC, NGS |
| Neuropatías hereditarias sensitivo-motoras | CMT | PCR, SEC, NGS |
| Paraplejías espásticas hereditarias | HSP | SEC, NGS |
| Ataxias hereditarias | HA | TP-PCR, SEC, NGS |
El diagnóstico definitivo de una ENM requiere de una cuidadosa evaluación clínica del paciente que oriente al profesional hacia cuál(es) gen(es) debe(n) ser estudiado(s) en el laboratorio de biología molecular. Una vez solicitado el estudio genético, el laboratorio de biología molecular (Tabla 1) deberá estudiar dicho gen (o genes) analizando si existe en el ADN del paciente, una mutación del tipo puntual, inserción, deleción, duplicación o expansión de repeticiones de nucleótidos (ver Glosario). Posteriormente, el profesional tratante deberá contar con el conocimiento y criterio necesarios que le permita reconocer si los resultados obtenidos en el laboratorio de biología molecular son consistentes con su impresión clínica y/o si tienen valor diagnóstico definitivo.
En los párrafos que siguen se presentan, de manera general y resumida, las estrategias de análisis de biología molecular de las ENM más frecuentes. Esta revisión está dirigida al neurólogo generalista y evita ahondar en la complejidad molecular de los estudios mencionados y/o en sus características técnicas o limitaciones mayores, ni considera las estrategias interactivas diagnósticas con estudios clínicos complementarios, tales como RMN de cerebro o músculos, evaluaciones electrofisiológicas y/o de anatomía-patológica. Además, a excepción de ciertas referencias seleccionadas relativas a ENM y/o su estudio a través de secuenciamiento masivo de ADN, se ha intentado citar revisiones generales, incluyendo los GeneReviews® [Internet] del NCBI, los cuales son fácilmente accesible a través de Internet. Esta última herramienta, junto al martillo del neurólogo, se ha convertido en un complemento esencial en el consultorio para la evaluación de pacientes con ENM.
DISTROFIA MUSCULAR DE DUCHENNE/BECKERLa distrofia muscular de Duchenne/Becker (DMD) es paradigmática entre las ENM dado que es la enfermedad muscular hereditaria más frecuente, tiene un patrón de herencia ligado al cromosoma X (Xp21) y el gen responsable (DMD), que codifica la proteína distrofina, fue el primer gen de una ENM descubierto6. La DMD afecta a varones, con una incidencia de ∼1:5000 varones nacidos vivos, manifestándose también clínicamente en ∼2.5-7.8% de las mujeres portadoras7. Las mutaciones responsables de la DMD consisten en la deleción de uno o más exones del gen DMD (60-70%), duplicaciones (5-10%) o mutaciones puntuales (25-35%), constituyendo estas últimas una fracción menor en la forma de Becker de la enfermedad (10-20%)6. Las diferentes mutaciones en el gen de distrofina impactan de manera variable en el grado de déficit muscular y/o el perfil cognitivo o compromiso cardíaco de los pacientes6. Algunas mutaciones, por ejemplo, dependiendo del grado de alteración de la estructura y/o función de la proteína distrofina resultante, conducen al cuadro clínico relativamente más benigno denominado distrofia muscular de Becker6. El estudio genético en pacientes con DMD permite:
- (i)
Llegar al diagnóstico de certeza de la enfermedad.
- (ii)
Estudiar mujeres a riesgo en la familia a fin de brindar un adecuado asesoramiento genético.
- (iii)
Reconocer la mutación específica en cada paciente a fin de disponer de eventuales elementos pronósticos (i.e., forma Duchenne versus Becker).
- (iv)
Ofrecer, eventualmente, un tratamiento personalizado y/o participación del paciente en protocolos farmacológicos específicos para distintos tipos de mutación. Inicialmente, el gold standard de los estudios genéticos para el gen DMD consistió en el análisis denominado PCR-multiplex (ver Glosario), en el que se analizan, simultáneamente, los exones más frecuentemente delecionados en pacientes con DMD8. Actualmente, la estrategia más utilizada es la de MLPA (ver Glosario), la cual analiza el total de los exones del gen DMD de manera cuantitativa. La estrategia de MLPA permite reconocer deleciones y duplicaciones de uno o más exones del gen DMD y estudiar, además, mujeres potencialmente portadoras de alelos de deleción o duplicación de este gen, permitiendo ofrecer así un adecuado asesoramiento genético familiar y/o diagnóstico en mujeres a riesgo con síntomas de déficit muscular. Finalmente, en aquellos casos en los que existe una fuerte sospecha clínica de DMD, siendo el estudio de MLPA normal (i.e., negativo para deleción o duplicación en el gen de distrofina), debe realizarse el análisis de secuencia completa del gen DMD en la búsqueda de mutaciones puntuales, las cuales no son reconocidas por MLPA.
La atrofia muscular espinal (AME) es la enfermedad hereditaria AR más frecuente siguiendo a la fibrosis quística o mucoviscidosis, con una incidencia de ∼4-10:100000 nacidos vivos y con una frecuencia de individuos portadores sanos en la población general de ∼1:50-90, dependiendo de factores étnicos9. Las formas clínicamente relevantes de AME, basadas en función de la severidad de aspectos motores específicos, son:
- (i)
AME tipo I o Werdnig-Hoffman: los pacientes no llegan a sentarse y generalmente fallecen antes de los dos años de vida;
- (ii)
AME tipo II o intermedia: los pacientes adquieren la sedestación, pero no la marcha;
- (iii)
AME tipo III o Kugelberg-Welander: los pacientes adquieren la marcha9.
Esta calificación simplificada y clínicamente práctica, no incluye subdivisiones adicionales de las formas tempranas o severas de AME ni la forma de AME adulta9. Una adecuada experiencia en la evaluación clínica de pacientes con hipotonía neonatal permite al neonatólogo o al neuropediatra reconocer clínicamente a los individuos AME tipo I y solicitar el estudio genético a fin de confirmar la sospecha clínica. En los casos de AME tipo II o III, la sospecha diagnóstica inicial puede ser más compleja dada la superposición clínica de estos pacientes con otras ENM9. En cualquier caso, AME I, II y III, están ocasionadas por mutación en un mismo gen, denominado SMN1 (gen de sobreviva de la neurona motora), localizado en la región cromosómica 5q1310. El gen SMN1 debe estudiarse con fines diagnósticos, mientras que un gen denominado SMN2, homólogo de SMN1 y localizado en la misma región cromosómica, tiene relativo valor pronóstico y relevancia en el tratamiento de AME11. La mutación más frecuentemente reconocida en el gen SMN1 es la deleción homocigota del gen10. El estudio genético habitual de pacientes AME I, II y III, consiste en el análisis de deleción homocigota de los exones 7 y 8 del gen SMN1, mediante la estrategia de PCR-RFLP12 o MLPA (ver Glosario)9. Es de destacar que el estudio de PCR-RFLP del gen SMN1 solo reconoce individuos con deleción homocigota (i.e., ambos alelos, materno y paterno, con deleción) y no individuos heterocigotas (i.e., un alelo con deleción y otro alelo portador de mutación puntual), los cuales constituyen ∼4% de los casos de AME13. Por este motivo, en caso de resultar normal el estudio de PCR-RFLP del gen SMN1 y sostenerse la sospecha clínica de AME, debe realizarse secuencialmente:
- (i)
El estudio cuantitativo del número de copias del gen SMN1, a través de PCR semicuantitativa, PCR de tiempo real o MLPA, a fin de reconocer la presencia de al menos un alelo portador de deleción y, en caso positivo.
- (ii)
Secuenciar el gen SMN1 en busca de la potencial mutación puntual en el otro alelo, que permita eventualmente confirmar el diagnóstico de AME9.
El estudio cuantitativo del número de copias del gen SMN1 también permite evaluar el estado de portador en individuos de la familia (padres, hermanos, tíos -paternos y maternos-) o en parejas que solicitan asesoramiento genético. En estos estudios debe considerarse la existencia de cromosomas portadores de dos copias del gen SMN1, en la población normal, que pueden llevar a falsos negativos en el estudio de portadores (i.e., individuos 2:0). En cualquier caso, el análisis adecuado de portadores en AME requiere de conocimiento y experiencia en estudios de biología molecular del gen SMN19.
DISTROFIA MIOTÓNICA DE STEINERTLa distrofia miotónica de Steinert (DM), segunda ENM en frecuencia, es una afección multisistémica, con herencia AD, en la que se han reconocido dos formas genéticas clínicamente similares, DM1 (19q13.32) y DM2 (3q21.3), asociadas a la expansión de las secuencias de tri y tetranucleótidos (CTG)n y (CCTG)n, respectivamente14,15. Los fenotipos clínicos de DM1, la forma más frecuente de DM, incluyen una forma congénita severa (no reconocida en DM215), formas infantiles sub-diagnosticadas, en las que predominan inicialmente alteraciones en aprendizaje, y formas juveniles y del adulto, con manifestaciones clínicas de déficit muscular y/o alteraciones cardiológicas y oftalmológicas (cataratas)14. Las bases moleculares de la DM fueron descubiertas en la década del 90’, casi simultáneamente con las de síndrome del cromosoma X-frágil y la atrofia muscular espinobulbar (SBMA; enfermedad de Kennedy). En estas patologías, así como en otras afecciones neurológicas reconocidas posteriormente, los genes alterados son portadores de una expansión anormal de repeticiones de nucleótidos16. La naturaleza de la mutación en DM1, consistente en una repetición inestable (CTG)n, en la región 3’UTR del gen DMPK, sugirió inmediatamente una explicación al fenómeno llamado de anticipación clínica observado en DM: expansiones adicionales de la secuencia (CTG)n, en generaciones sucesivas, da lugar a fenotipos clínicos más severos y de inicio a edad más temprana, siendo la forma congénita de la DM la manifestación más grave de la enfermedad14. El análisis genético de DM1, realizado en los laboratorios generales de biología molecular, consiste en la determinación del tamaño de ambos alelos (CTG)n (provenientes del padre y la madre del paciente) a través de las metodologías de PCR y/o TP-PCR17 (ver Glosario). Recientemente se han observado pacientes DM1 con expansión de repeticiones CTG interrumpidas por trinucleótidos CCG o CTC, las cuales pueden conducir a falsos resultados negativos en el análisis de TP-PCR18. Esta situación diagnóstica es solucionada utilizando dos reacciones de TP-PCR, una en el extremo 5’ y la otra en el extremo 3’ de la repetición (CTG)n, de manera que al menos una reacción detecte la eventual expansión18.
DISTROFIA MUSCULAR FACIOESCAPULOHUMERAL (FSHD)FSHD es una ENM con marcada variabilidad en su presentación clínica, progresión y edad de inicio, reconociéndose formas clínicas infantiles severas y formas juveniles o adultas, eventualmente más moderadas19. Esta afección, clínicamente sub-diagnosticada, representa junto a la distrofia miotónica de Steinert la segunda ENM en frecuencia20. Las bases moleculares de la FSHD son complejas e incluyen elementos genéticos y epigenéticos, habiéndose reconocido dos formas genéticas principales FSHD1 y FSHD2, clínicamente idénticas21. La forma más frecuente (FSHD1) está ligada a la región subtelomérica del brazo largo del cromosoma 4 (región 4q35) donde existe una repetición en tándem polimórfica de un número variable de unidades de 3,3 Kb (entre 10 y 100, en la población normal) denominadas D4Z4. Los pacientes FSHD1 presentan un acortamiento en el número de estas unidades, presentando alelos de 1-10 D4Z4, asociado a una disminución en el grado de metilación de los nucleótidos citosinas en las unidades D4Z4 remanentes y a la presencia de una región denominada 4qA (haplotipo permisivo). Los pacientes FSHD2, por otra parte, representan menos de un 5% de los individuos FSHD y son portadores de mutaciones en el gen SMCHD1 o, en menor medida, otros genes19, e incluyen dos elementos comunes con FSHD1: la disminución del grado de metilación de citosinas en el tándem D4Z4 y el haplotipo 4qA19,22. Frente a la sospecha clínica de FSHD, el estudio genético diagnóstico habitual consiste en el análisis del número de unidades D4Z4 en 4q35, realizado a través de PFGE y SB (ver Glosario) a fin de establecer si existe un acortamiento de este tándem y, eventualmente, confirmar así el diagnóstico clínico. La ausencia de acortamiento del tándem requiere de una evaluación correcta de las limitaciones de la metodología usada para este estudio y, eventualmente, del análisis de secuencia completa y de del/dup (ver Glosario) del gen SMCHD1.
DISTROFIAS MUSCULARES DE CINTURASLas distrofias musculares de cinturas (Limb Girdle Muscular Dystrophy o LGMD) comprenden un extenso grupo de ENM con elementos semiológicos comunes, vinculados a su nombre genérico (i.e., debilidad y amiotrofia progresiva de grupos musculares de las cinturas escapular y pelviana) con diferencias fenotípicas clínicas importantes en cuanto a edad de inicio, progresión de la enfermedad, patrón de músculos afectados, alteraciones osteoarticulares, así como neumológicas y cardiológicas23. Existe una marcada heterogeneidad genética en las formas autosómicas dominantes (LDMD1) y recesivas (LGMD2) de las LGMD23 por lo que el estudio de un gen en particular ha conducido frecuentemente a resultados negativos. El análisis secuencial gen-tras-gen en pacientes con LGMD está siendo reemplazado24 por el estudio simultáneo de múltiples genes (i.e., paneles de genes y/o análisis de exoma o WES) (ver Glosario), acelerando así los tiempos diagnósticos en los pacientes en este grupo de ENM.
MIOPATÍAS DISTALES Y MIOPATÍAS MIOFIBRILARESEstas entidades representan dos grandes grupos de ENM, poco frecuentes y genéticamente heterogéneas, con características clínicas comunes que incluyen una forma de herencia AD y afectación inicial distal de miembros inferiores con inicio en la edad adulta. Algunas entidades clásicamente llamadas miopatías distales (i.e., desminopatías, miotilinopatías distales, ZASPopatías y las originadas por mutaciones en α-B-cristalina) muestran cambios anatomo-pa tológicos en el músculo similares a los observados en las llamadas miopatías miofibrilares, las cuales son todas ocasionadas por mutaciones en genes de proteínas de la línea Z del sarcómero25. Si bien, como en la gran mayoría de las ENM, la sospecha diagnóstica de estos pacientes requiere de un adecuado abordaje clínico, anatomo-patológico y eventualmente estudio de RMN de músculo, el diagnóstico genético definitivo está crecientemente asociado al estudio a través de paneles de genes y/o WES25.
MIOPATÍAS CONGÉNITAS Y DISTROFIAS MUSCULARES CONGÉNITASEstos dos grupos complejos de ENM han sido diferenciados históricamente por su presentación clínica y por hallazgos morfológicos y estructurales en el análisis del músculo27. Las distrofias musculares congénitas (CMD) son afecciones musculares de inicio temprano en la vida con evidencia de distrofia en la evaluación anatomo-patológica del músculo26, mientras que las miopatías congénitas (CM) presentan características histopatológicas y ultraestructurales diferenciales [i.e., rods - miopatía nemalínica- cores (central core disease o CCD), núcleos centrales (miopatías centronucleares o CNM)], con o sin distrofia27. Recientes hallazgos clínicos y genéticos, fundamentalmente basados en el estudio de un número creciente de pacientes, han ampliado el espectro de fenotipos clínicos y su superposición eventual entre las CMD, las CM e, incluso, las LGMD27. Si bien la evaluación clínica y los estudios anatomo-patológicos siguen siendo relevantes para la sospecha diagnóstica en estas ENM, el estudio simultáneo del conjunto de genes causales de todas las formas reconocidas de CMD y CM, a través de un panel de genes, permite una rápida y completa evaluación genética de estos pacientes. Los resultados obtenidos, incluyendo potenciales mutaciones patogénicas y VUS (ve Glosario), deben cotejarse cuidadosamente con las observaciones clínicas28 (ver Conclusión).
DISTROFIA MUSCULAR OCULOFARÍNGEALa distrofia muscular oculofaríngea es una ENM aparentemente sub-diagnosticada que se trasmite fundamentalmente con un modo de herencia AD29. La afección tiene inicio en la edad adulta y es caracterizada por ptosis palpebral progresiva, disfagia, debilidad proximal muscular en miembros y por la aparición de un tipo particular de inclusiones intranucleares en la fibra muscular29. La mutación causal es una expansión patológica corta de la secuencia trinucleotídica (GCG)n en el gen PABPN1 (polyadenylate-binding protein nuclear 1 gene) localizado en la región cromosómica 14q11.1. El estudio diagnóstico de biología molecular se realiza por PCR y/o secuencia de Sanger (ver Glosario).
SINDROMES MIASTÉNICOS CONGÉNITOSLos sindromes miasténicos congénitos son un grupo de ENM poco frecuentes, que resultan de alteraciones en la trasmisión neuromuscular30. Los genes alterados en estos pacientes codifican proteínas localizadas en la sinapsis neuromuscular31. El diagnóstico clínico está basado fundamentalmente en el hallazgo de fatigabilidad y/o debilidad muscular fluctuante que afecta especialmente a los músculos oculares y otros músculos craneales, con antecedentes familiares positivos y un perfil acorde en los estudios electrofisiológicos30. Actualmente el camino diagnóstico genético más eficiente está basado en el estudio de un panel de genes específicos31.
NEUROPATÍAS HEREDITARIAS SENSITIVO-MOTORASLas neuropatías hereditarias sensitivo-motoras (HSMN) o enfermedad de Charcot-Marie-Tooth (CMT) presentan heterogeneidad clínica y una marcada heterogeneidad genética, con patrones de herencia AD, AR o ligada al X, asociadas a defectos en múltiples genes32. Una adecuada inspección clínica neurológica, evaluación de edad de inicio y progresión de la afección, antecedentes familiares y modo de herencia, así como el resultado de estudios complementarios (EMG, velocidades de conducción, biopsia de nervio) y diagnóstico diferencial con formas adquiridas de HSMN (inflamatorias o inmunomediadas -CIDP-, infecciosas, déficit vitamina B12, etc.), puede orientar hacia un estudio genético apropiado para CMT33. Las formas clínicas más frecuentes se denominan CMT1 (∼50%), representando la variante CMT1-A el 70-80% de las mismas. Por este motivo, el algoritmo diagnóstico en CMT comienza generalmente con el análisis de CMT1-A, cuya mutación más frecuente está asociada a una microduplicación del gen PMP22, en la región 17p11.2, y a la presencia patológica de tres copias de dicho gen. El estudio convencional de microduplicación de PMP22 se realiza por análisis de PCR semicuantitativa de alelos de marcadores microsatélites polimórficos en la región 17p11.2. Mutaciones puntuales en el gen PMP22 también son causales de CMT (denominada CMT1-E) y deben estudiarse a través del análisis de secuencia y del/dup de dicho gen. La deleción de una copia del gen PMP22 conduce a otra forma de HSMN denominada HNPP (Hereditary Neuropathy with Liability to Pressure Palsy), utilizándose para el diagnóstico de biología molecular de HNPP la misma estrategia de análisis cuantitativo de alelos usada para microduplicación en CMT1-A. Actualmente se dispone también del estudio de MLPA para el análisis de duplicaciones o deleciones en la región del gen PMP22. En aquellos casos donde la sospecha clínica no está orientada hacia CMT1-A, o que el estudio del gen PMP22ha sido negativo, existen algoritmos diagnósticos que pueden orientar hacia el estudio de otros genes vinculados a distintas formas de CMT (i.e., PMZ, GJB1, GDAP1, entre otros)33. Sin embargo, en individuos negativos para microduplicación CMT1-A, el estudio de un panel de genes CMT, con análisis de del/dup, puede ser más eficiente que la estrategia gen-tras-gene.
PARAPLEJÍAS ESPÁSTICAS HEREDITARIASLas paraplejías espásticas hereditarias (HSP), ya sea en sus formas puras o complejas (por ejemplo, asociadas a ataxia), constituyen un gran grupo de afecciones neurológicas con una prevalencia global estimada de 1-5:100000 y cuyos signos relevantes son axonopatía distal y espasticidad progresivas34. Las HSP son trasmitidas a través de diversas formas de herencia, estando asociadas a una gran heterogeneidad genética que incluyen más de 50 genes (http://www.musclegenetable.fr/). Los genes más frecuentemente afectados en las formas AD de HSP son SPAST, ATL1, REEP1 y KIF5A, mientras que los genes SPG11, CYP7B1, SPG7 y SPG15 están afectados más frecuentemente en las formas AR35. La disponibilidad actual de paneles de genes para estudio de HSP, así como para el estudio combinado con paneles de ataxias hereditarias, permite eventualmente acceder más rápidamente al diagnóstico genético definitivo de estos pacientes35.
ATAXIAS HEREDITARIASAunque no son incluidas habitualmente en el grupo específico de las ENM, las ataxias hereditarias constituyen un grupo relevante de afecciones neurológicas de observación frecuente en la práctica del neurólogo clínico (http://www.musclegenetable.fr/). Estas afecciones incluyen un gran número de formas esporádicas, sin antecedentes familiares, y otras que muestran patrones de herencia AD, AR, ligadas al cromosoma X o de herencia mitocondrial36. El algoritmo diagnóstico del neurólogo clínico para las ataxias hereditarias, previo a la solicitud de todo estudio de biología molecular, tiene gran relevancia dada la gran variedad de genes posibles afectados y fundamentalmente por la necesidad de un diagnóstico diferencial con formas adquiridas de ataxia, no hereditarias37. Entre las formas relevantes de ataxias hereditarias AD se encuentran el gran grupo de las ataxias espinocerebelosas (SCAs), para las cuales existen fenotipos clínicos orientadores36,37 y paneles de genes para su estudio. Entre las formas clínicas con herencia AR, se destaca la ataxia de Friedreich (AF). Esta afección es causada por la expansión patológica de una secuencia de trinucleótidos (GAA)n, en el primer intrón del gen FXN (región cromosómica 9q21.11) que codifica para la proteína frataxina. El estudio de biología molecular de la AF consiste en la caracterización del tamaño de los alelos (GAA)n en el gen de frataxina, a través de las metodologías de PCR y TP-PCR.
MIOPATÍAS METABÓLICAS, CANALOPATÍAS, PARÁLISIS PERIÓDICAS FAMILIARES Y MIOPATÍAS MITOCONDRIALESLa descripción de los estudios de biología molecular para las entidades incluidas en estos grupos de ENM excede el marco de este artículo. Entre estas, sin embargo, debe destacarse el impacto clínico del estudio enzimático y eventualmente genético para la enfermedad de Pompe (glucogenosis tipo II), el cual es accesible en la mayoría de los laboratorios de bioquímica o biología molecular y para cuyos pacientes existe un tratamiento de sustitución enzimática curativo. En el grupo de las afecciones de origen mitocondrial, actualmente es posible estudiar el genoma mitocondrial completo y/o paneles de genes nucleares con impacto en la función mitocondrial38.
ESTUDIOS GENÉTICOS PRE-SINTOMÁTICOSEl estudio genético pre-sintomático se realiza a un individuo sano, a riesgo de padecer una enfermedad neurológica en su vida adulta39 Este tipo de estudio ha sido erróneamente llamado “diagnóstico” pre-sintomático (i.e., del inglés, “pre-symptomatic testing”) dado que, no habiendo signos y síntomas de enfermedad en un individuo sano, no hay posibilidad “diagnóstica” alguna. Si bien la afección neurológica paradigmática en este grupo es la enfermedad de Huntington, en el conjunto de otras afecciones (i.e., demencias frontotemporales, formas familiares de Alzheimer y de la enfermedad de Parkinson, etc.), estos estudios pueden realizarse también para algunas ENM (i.e., DM, FSHD, DMOP, CMT, SCAs, HSP, formas familiares de ELA y laminopatías). Los beneficiarios de estos estudios son generalmente hijos o nietos de individuos afectados, quienes concurren al médico genetista o al neurólogo para consultar acerca del riesgo de padecer ellos mismos la enfermedad en el futuro y/o de trasmitirla a su descendencia39. En la mayoría de estas situaciones, el individuo realiza la consulta con una importante impronta emocional frente a la enfermedad en cuestión, dado que ha conocido y/o presenciado, en mayor o menor detalle, los pormenores de los inicios y/o progresión de la misma en un familiar cercano. Además, a pesar de su genuino y comprensible deseo de conocer su potencial status de portador, no necesariamente está preparado psicológicamente para enfrentarse al resultado de este análisis, ya sea el mismo negativo o positivo para la mutación en estudio. Esta delicada situación bioética y psicológica requiere de una evaluación y preparación previa del individuo de manera que, eventualmente, pueda integrar su nueva dimensión del futuro, diferenciar entre tiempo de transición en salud versus espera de la enfermedad o, en ciertos casos, renunciar a la realización del estudio por anticipar sus consecuencias39. En familias con hermanos portadores y no portadores de un gen afectado, por ejemplo, puede resentirse el sentimiento de identidad de grupo al vislumbrar que unos tienen un destino diferente de otros, creando una trágica disimetría en la familia39. Por estos motivos, es recomendable una cuidadosa atención, tanto en las fases previas como en la entrega del resultado y posteriores, de todo aquel individuo sano que, siendo mayor de edad como requisito, solicite un estudio genético pre-sintomático. El resultado de este estudio debe darse siempre y exclusivamente en una consulta cara a cara y con la previsión adecuada de la atención futura del individuo.
CONCLUSIÓNCiertas formas frecuentes y clínicamente estereotipadas de ENM, tales como la distrofia muscular de Duchenne (DMD), la atrofia muscular espinal (AME) o la distrofia miotónica de Steinert (MD), son habitualmente estudiadas a través del análisis directo de los genes responsables (i.e., distrofina, SMN1 y DMPK, respectivamente). Sin embargo, en muchas otras formas clínicas de ENM (i.e., distrofias de cinturas, neuropatías periféricas, miopatías congénitas y otras), el estudio de un gen particular puede conducir a un resultado negativo que requiere del análisis secuencial de un posterior gran número de genes los que, a su vez, pueden resultar todos normales. Esta situación de análisis gen-tras-gen se ha modificado dramáticamente gracias a los modernos métodos de estudio simultáneo de múltiples genes (i.e., paneles de genes y/o análisis de exoma), acelerando así los tiempos diagnósticos y disminuyendo las consecuencias emocionales en el paciente y los costos vinculados al estudio gen-tras-gen. Como consecuencia del análisis de paneles de genes, WES o WGS, sin embargo, aparece una frecuente complicación diagnóstica, asociada al reconocimiento de variantes genéticas para las que se desconoce su significado clínico (variantes VUS). El médico debe interpretar adecuadamente el significado de estos resultados, caso contrario puede ofrecer un diagnóstico equivocado al paciente. En la mayoría de los casos, sin embargo, los estudios de biología molecular de gen único o genes múltiples, en pacientes con ENM, ofrecen al paciente y su familia un diagnóstico genético de certeza, una expectativa de pronóstico más acertado, pautas de tratamiento adecuadas, un correcto asesoramiento genético familiar, así como la potencial participación en eventuales protocolos clínicos farmacológicos y/o tratamientos específicos y/o personalizados.
Alelos: Formas alternativas de un gen en un mismo locus.
Cobertura: Número independiente de veces en que un determinado nucleótido es “leído”. Mientras mayor es este número (50x, 100x, etc.) la secuencia es más confiable. En ciertas regiones del genoma humano la cobertura puede no ser óptima.
Deleción: Pérdida de uno o más nucleótidos, incluso de grandes fragmentos de ADN.
Duplicación: Repetición en tándem de un nucleótido, o de una secuencia de nucleótidos, incluso de grandes fragmentos de ADN.
Del/Dup: Análisis de la presencia de deleción o duplicación en un gen.
Exoma: Conjunto de exones de todos los genes humanos.
Gen: Unidad física y funcional genética que ocupa una posición definida en el genoma y contiene la información (codificada) de la secuencia de aminoácidos de una o más proteínas.
Genoma: Material genético completo de un individuo, incluyendo regiones exónicas y no exónicas, así como el genoma mitocondrial.
Heterocigota: Presencia de dos alelos diferentes, en cada cromosoma, para un dado gen.
Homocigota: Presencia de dos alelos idénticos, en cada cromosoma, para un dado gen Inserción: Incorporación aberrante de un nucleótido, o de una secuencia de nucleótidos, incluso de grandes fragmentos de ADN.
Inversión: Aberración cromosómica en la que se invierte el orden de una secuencia de nucleótidos, incluso de grandes fragmentos de ADN.
MLPA (Multiplex Ligation-dependent Probe Amplification): Técnica de biología molecular que permite reconocer deleciones y duplicaciones a través de un conjunto de sondas, específicas para los distintos exones o regiones de un dado gen, las cuales son unidas y amplificadas por PCR para posterior análisis cuantitativo de los productos obtenidos.
Mutación: Todo cambio en la secuencia del ADN.
Mutación puntual: Cambio en la secuencia de un nucleótido.
Panel de genes: Estudio de un conjunto de genes vinculados a una entidad clínica o entidades clínicas relacionadas. La cobertura obtenida en los estudios de paneles de genes es mayor que en WES o WGS. Ciertos genes, de reciente análisis o asociación al fenotipo clínico en estudio, pueden no estar incluidos en paneles de genes no actualizados.
PCR (Polymerase chain Reaction): Técnica de amplificación de un fragmento de ADN usando una ADN-polimerasa termoestable.
PCR-RFLP: Amplificación de un fragmento de ADN por PCR seguida de digestión del mismo con una o más endonucleasas de restricción y análisis de los fragmentos resultantes.
SEC (secuenciamiento de Sanger): Metodología de secuenciación directa del ADN, por lo cual no existen limitaciones de cobertura.
SB (Southern blot): Estudio de moléculas de ADN transferidas a un soporte sólido (membrana de nylon o nitrocelulosa) usando sondas especificas.
Secuenciación del ADN: Determinación de la secuencia de nucleótidos del ADN.
Secuenciación masiva de ADN o NGS (Next Generation Sequencing): Permite analizar un gen, múltiples genes en forma simultánea, o el exoma (WES) o genoma (WGS), de un individuo. La secuenciación masiva se realiza usando tecnologías sofisticadas bioquímicas que permiten obtener “lecturas” (reads) para posterior análisis bioinformático (i.e., alineamiento de lecturas respecto a un genoma de referencia, asignación de nucleótidos, mapeo, análisis de variantes y anotación). Las variantes identificadas en cada gen son filtradas según su frecuencia alélica e impacto teórico funcional según predicciones de algoritmos in silico. Las variantes relevantes se evalúan usando bases de datos, tanto de mutaciones pertinentes a enfermedades específicas como de polimorfismos (SNPs) generales. El proceso de análisis de variantes genéticas es un proceso minucioso que requiere de tiempo y experiencia. Si bien existen lineamientos generales para la interpretación de variantes, el potencial patogénico de una dada variante puede permanecer incierto.
TP-PCR (Triplet repeat Primed-PCR): Técnica de PCR que permite amplificar eficientemente secuencias de ADN portadoras de repeticiones de trinucleótidos.
VUS (Variants of Unknown Significance): Cambios en la secuencia del ADN, encontrados en el estudio de secuenciación de genes, para los que se desconoce su rol patogénico.
WES (Whole Exome Sequencing): Estudio de los exones de todos los genes humanos, los cuales representan ∼2% del genoma, comprenden ∼85% de las mutaciones causales de enfermedades monogénicas conocidas, y han sido de utilidad diagnóstica en aproximadamente el 29-40% de los casos analizados de ENM40. La información obtenida en WES, aún luego del filtrado, incluye múltiples VUS que pueden dificultar el análisis, situación eventualmente subsanable a través de la evaluación de tríos (caso índice, madre y padre). El análisis de tríos permite determinar el origen, eventualmente paterno y/o materno, de una o más variantes de tal manera que: i) en el caso de enfermedades AR, es posible establecer si dos variantes potencialmente causales de enfermedad están en configuración cis o trans; ii) es posible identificar mutaciones de novo en enfermedades autosómicas dominantes. El uso de WES con fines clínicos tiene aún limitaciones, vinculadas fundamentalmente a la cobertura insuficiente de ciertos exones y a su alto costo.
GS (Whole Genome Sequencing): Estudio del genoma completo de un individuo que incluye también regiones intergénicas y rearreglos cromosómicos no detectables por MLPA ni observables en estudios de DNA-microarrays (no comentados en esta revisión). En los WGS existen regiones con cobertura incompleta y resultados de baja reproducibilidad para variantes de inserción/deleción. La utilidad diagnóstica en ENM ha sido recientemente evaluada40.
Este trabajo ha sido financiado por subsidios de FONCYT PICT-2015-1581 (Argentina) y de la FSH Society (EE.UU.; Grant FSHS-82015-03). SP es Becaria de Doctorado de FONCYT. ALR es Investigador Principal de CONICET.