






La hipotonía es un signo no específico definido como una disminución de la resistencia al movimiento pasivo de las articulaciones, la cual comprende un grupo amplio y heterogéneo de condiciones que afectan tanto al sistema nervioso central como periférico. En la orientación clínica sigue siendo crucial distinguir pacientes con o sin debilidad asociada y la diferenciación entre hipotonía central, periférica y mixta. A través de la historia clínica y examen físico es posible lograr esta distinción en cerca de la mitad de los casos, siendo las centrales las más frecuentes. Este artículo tiene como objetivo realizar una puesta al día en síndrome hipotónico del recién nacido y lactante menor (<2 años), con énfasis en las causas de origen neuromuscular hereditario, fundamentalmente en la evaluación clínica y enfoque diagnóstico.
Hypotonia is a non-specific sign defined as decrease resistence to passive joint's movement, wich comprises a broad and heterogeneous group of conditions that affect the central or peripheral nervous system. in the first clinical orientation it is still crucial to distinguish patients with oy without associated weakness and the differentiation between central, peripheral and mixed hypotonia. Through clinical history and physical examination it is possible to achieve this distinction in about half of the cases, with central causes being the most frequent. The objetive of this article is to make an update hypotonic syndrome in newborns and infants (less than 2 years), with emphasis on hereditary neuromuscular causes, focused on clinical evaluation and diagnosis approach.
La hipotonía es un signo no específico que clásicamente se ha definido como la disminución de la resistencia al movimiento pasivo de las articulaciones, llevando a un incremento excesivo en los rangos de movimiento articular1. En los neonatos e infancia temprana, la alteración de la postura secundaria a la hipotonía es lo más llamativo, sin embargo en infancia tardía o niñez, esta puede manifestarse sólo como retraso en la adquisición de hitos del desarrollo motor, por lo que a esta edad generalmente ya no se habla de hipotonía como diagnóstico sindromático principal. La diferenciación entre hipotonía y debilidad muscular sigue siendo crucial en la aproximación diagnóstica, siendo esta última definida como la disminución de la máxima fuerza que puede ser generada, hecho que divide clásicamente a pacientes hipotónicos “paralíticos” (hipotonía periférica) de “no paralíticos” (hipotonía central)2.
La incidencia y prevalencia del sindrome hipotónico es difícil de determinar, dado que es una característica presente en muchos trastornos de diversa etiología3, sin embargo es un motivo de consulta frecuente en pediatría, de hecho algunos estudios en nuestro medio muestran que hasta representan un 5% de los motivos de ingreso a neonatología4.
Si bien la hipotonía es un signo, la mayoría de las veces fácilmente reconocible, encontrar la causa puede llegar a ser un gran desafío diagnóstico para el clínico. Las razones son múltiples, de hecho puede observarse en forma fisiológica en los recién nacidos prematuros, puede ser una manifestación de una enfermedad aguda en neonatos o lactantes sanos, o por el contrario, ser una manifestación de una enfermedad del sistema nervioso central y/o una enfermedad neuromuscular2. Si bien dentro de las principales causas están las de origen en el sistema nervioso central, las enfermedades originadas en el sistema nervioso periférico han ido cobrando importancia en la medida que se han desarrollado nuevas técnicas diagnósticas y terapéuticas, entre las cuales cabe destacar técnicas de electrofisiología, biopsia muscular y de piel, imágenes musculares y estudios genéticos, entre otros.
El objetivo de este artículo es hacer una puesta al día en sindrome hipotónico del recién nacido y lactante menor (<2 años), con énfasis en las causas de origen neuromuscular hereditario, fundamentalmente en la evaluación clínica que permitan identificar signos específicos que orienten en la solicitud de nuevas herramientas diagnósticas.
EVALUACIÓN CLÍNICADesde la década del 80, se ha postulado que tanto los antecedentes como el examen clínico de los pacientes con sindrome hipotónico, son los elementos más útiles para realizar un diagnóstico etiológico, llegando al diagnóstico hasta en un 50% de los recién nacidos y lactantes, con dichas herramientas2,5,6.
Historia ClínicaLa importancia de realizar una cuidadosa historia clínica en la evaluación de un niño hipotónico es muchas veces pasada por alto7. Se debe consultar sobre patologías del embarazo tales como el antecedente de reducción de movimientos fetales y la presencia de oligo y/o polihidroamnios, las cuales son altamente específicas de enfermedades neuromusculares (88.6 y 75%, respectivamente)8. También realizar un pedigree familiar lo más completo posible, de al menos tres generaciones, indagando en consanguinidad, patologías cardíacas y abortos a repetición, antecedentes que nos orientan a causas de origen genético y/o metabólico3. Se debe consultar dirigidamente sobre síntomas y signos de patologías neuromusculares en la familia, tales como fatigabilidad, hipertermia maligna, pie cavo o miotonías, que generalmente no son relatados espontáneamente. La forma de inicio de la hipotonía, si su instalación fue aguda, subaguda o crónica, la presencia de fluctuaciones y si es de curso estático o progresivo, orientan a distintas causas. A modo de ejemplo, en un recién nacido sano que desarrolla hipotonía aguda a las 12-24 horas de vida, una de las hipótesis a descartar debido al curso de ésta, es un error innato del metabolismo. Ver tabla 1.
Aspectos relevantes a considerar en historia clínica de paciente hipotónico
| Historia | Características |
|---|---|
| PRENATAL | |
| Maternas: | • Infección materna (Rubéola, CMV, Herpes simple, entre otros) |
| • Exposición a teratógenos | |
| • Patologías maternas previas (Diabetes mellitus, epilepsia, lupus, hiperlaxitud articular, miastenia, miotonía, entre otros) | |
| • Antecedentes de abortos a repetición | |
| • Edad avanzada materna (aneuploidía) | |
| • Diabetes gestacional | |
| PERINATAL | |
| Parto: | • Presentación anómala |
| • Parto múltiple | |
| • Trauma durante parto | |
| • Infección materna al momento del parto | |
| • Anestesia materna | |
| Neonatal: | • Hipoxia |
| • Alteraciones metabólicas (ej: acidosis, hiperamonemia, hipoglicemia, entre otros.) | |
| • Alteración succión/deglución | |
| • Necesidad de apoyo ventilatorio | |
| FAMILIAR | |
| • Antecedentes de patologías neuromusculares | |
| • Edad paterna avanzada (aumento de mutaciones de novo) | |
| • Consanguinidad | |
| • Muertes de lactantes | |
(REF. 7,26).
Al enfrentarnos a un paciente con sindrome hipotónico el examen físico general debe ser minucioso y completo, consignando dismorfias, anomalías cardíacas, presencia de insuficiencia respiratoria y visceromegalia (hepatomegalia). Desde el punto de vista motor, debe evaluarse fuerza, reflejos osteotendíneos, masa muscular (trofismo), reflejos arcaicos, sensibilidad y buscar signos específicos, tales como la presencia de miotonía (relajación muscular lenta luego de contracción voluntaria o percusión en eminencia tenar o lengua) y fasciculaciones linguales7. Todos estos aspectos son de especial relevancia en la localización de la lesión, central o periférica9. También es necesario evaluar a los padres, buscando miotonía o presencia de debilidad facial, las cuales orientan hacia una distrofia miotónica, mientras que el hallazgo de pie cavo y atrofia distal de extremidades inferiores, a una posible polineuropatía1,3,10,11.
Evaluación del tonoLa evaluación del tono muchas veces no es fácil, en especial para un examinador inexpérto, ya que se debe examinar a una gran cantidad de niños sanos para poder reconocer cambios sutiles12. Por otro lado, la evaluación depende de muchas variables, a modo de ejemplo, en unidades de neonatología es difícil realizar un primer examen confiable, ya que el tono puede verse afectado tanto por el nivel de vigilia del lactante, ingesta alimentaria reciente, edad gestacional, fármacos y enfermedades sistémicas intercurrentes, por lo que necesitaremos realizar evaluaciones seriadas para tener una visión más fidedigna12.
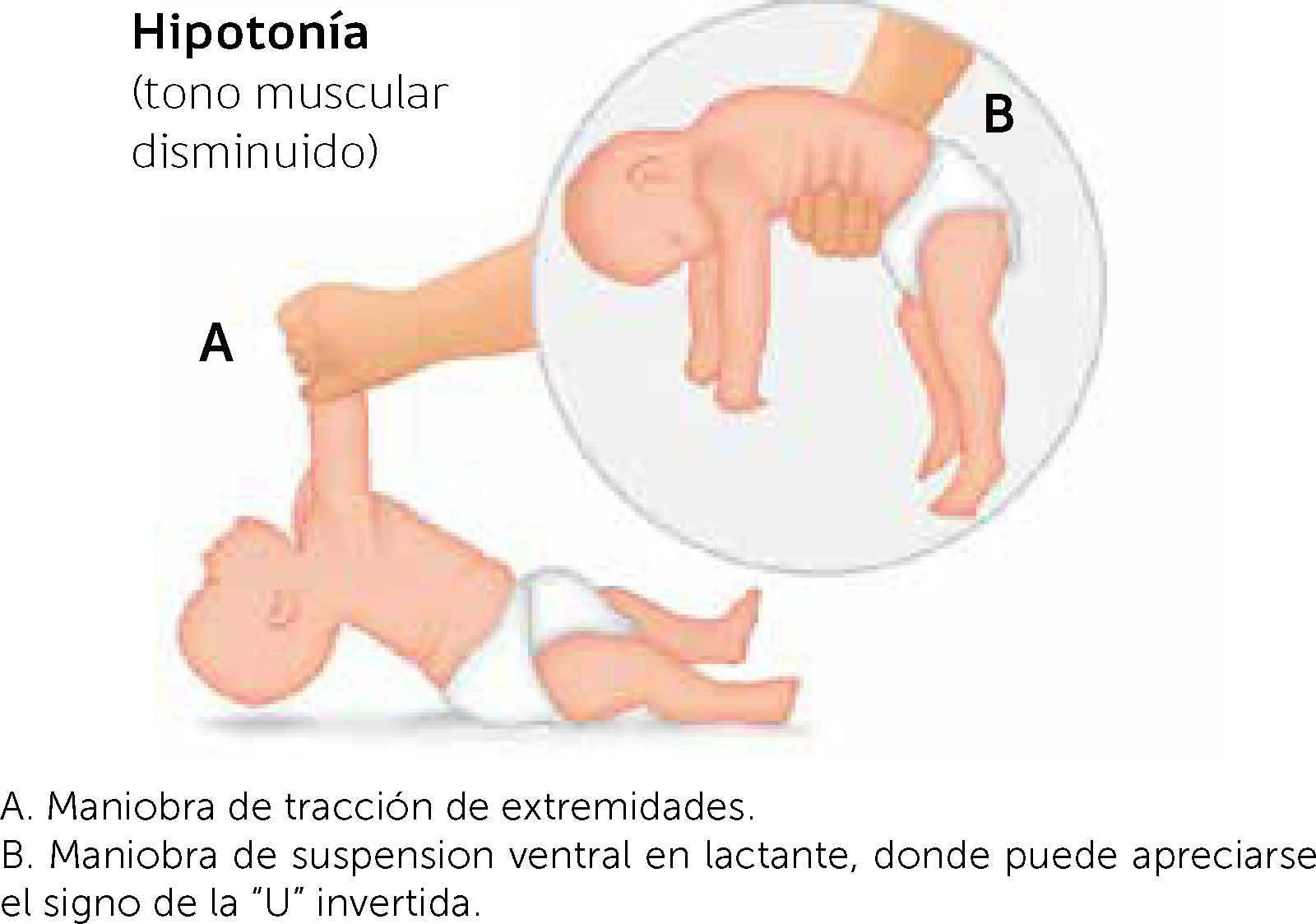
El primer paso de la evaluación es la observación de la postura espontánea, donde el paciente hipotónico estando en decúbito supino, se puede presentar una postura en batracio, esto es, caderas abducidas, extremidades inferiores en rotación externa y extensión flácida de brazos9. Existen diversas maniobras para evaluar tono en los lactantes, dentro de las cuales tenemos: tracción de extremidades superiores, suspensión vertical y suspensión ventral.La tracción de extremidades superiores evalúa tanto el tono axial, como debilidad de las extremidades superiores. El lactante en supino es traccionado desde las manos a sedente observando falta de control cefálico con rezago de la cabeza. En recién nacidos de término, hasta aproximadamente los 2 meses, lo normal es tener un ligero receso de la cabeza con respecto a la línea media11,12. La maniobra de suspensión vertical se realiza sosteniendo al lactante por debajo de los hombros en posición vertical. Se debe encontrar resistencia por parte del paciente y no deslizarse entre las manos del examinador. Este signo evidencia hipotonía axial y de cintura escapular12. En lactantes con afecciones a nivel central puede verse además, tendencia a la hiperextensión de extremidades inferiores, con entrecruzamiento de ellas durante esta maniobra12. La maniobra de suspensión ventral se realiza tomando al lactante por el dorso en posición supina, siendo lo esperado que sea capaz de mantener las extremidades en flexión y su cabeza más alta que la horizontal. Cuando nos enfrentamos a una hipotonía, el signo clásico se describe como en posición de U invertida11. Figura 1
Evaluación de Fuerza
La fuerza en niños mayores puede evaluarse fácilmente a través de escalas (Medical Research Council Scale)13, sin embargo no son aplicables a neonatos ni lactantes. En este segmento etario es de utilidad la observación de los movimientos antigravitacionales, tanto espontáneos, como frente a estimulación (ej: táctil). Ambos están disminuidos en pacientes con debilidad. Pueden ser clasificados según severidad en ausentes (no existe movimiento), severamente reducidos (sólo esbozo de movimiento) y parcialmente reducidos (algún movimiento, pero no completamente antigravitatorio)8. La fuerza también puede ser evaluada observando el llanto, succión, expresión facial, fuerza contra resistencia y esfuerzo respiratorio. Es importante también consignar la distribución de esta debilidad, si es proximal (ej: cintura escapular, pelviana), distal (ej: manos, pies) y si existe compromiso facial (ej: falta de mímica facial, fascie alargada)7,11.
Reflejos Osteotendíneos (ROT)La evaluación de los ROT puede orientar en el diagnóstico diferencial, estando ausentes en pacientes con afecciones de segunda motoneurona o nervio periférico y presentes y/o exaltados en afecciones del sistema nervioso central. Deben buscarse reflejos maseterino, bicipital, braquiorradial, rotuliano y aquiliano. Los signos de hiperreflexia son: aumento de área reflexógena, difusión y clonus, sin embargo, hay que tener en cuenta que el clonus hasta 10 batidas es esperable en lactantes menores de tres meses, que el reflejo plantar extensor puede verse hasta el año y que hasta un 10% de la población tiene difusión del reflejo rotuliano, siendo considerado normal hasta los 8 meses, siempre que no se acompañe de otra focalidad neurológica7.
CLASIFICACIÓN DE SINDROMES HIPOTÓNICOSLa primera distinción que debe realizarse frente a un lactante con hipotonía es si es secundaria a una afección a nivel del sistema nervioso central o del sistema nervioso periférico, es decir el primer paso es intentar localizar. Si realizamos una diferenciación clara, se puede llegar al diagnóstico etiológico en un 67-85% de los pacientes5. Las condiciones que afectan al sistema nervioso central, es decir a las vías supraespinales que comprenden las vías piramidales, extrapiramidales y cerebelosas, causan un cuadro clínico conocido como hipotonía central. Mientras que la hipotonía de origen periférico es secundaria a compromiso de algún componente de la unidad motora, esto es, desde el cuerpo de la motoneurona ubicado en el asta anterior de la médula, hasta la fibra muscular. En ocasiones no es tan simple realizar una distinción clara, ya que se superponen características de ambos cuadros, lo cual puede verse en patologías que afectan tanto sistema nervioso central como periférico, a lo cual denominamos sindrome hipotónico mixto. Esto se presenta, por ejemplo, en pacientes con distrofia muscular congénita, que pueden presentar malformaciones cerebrales asociadas, como también en pacientes con enfermedades neuromusculares que sufren alguna injuria aguda en el período neonatal (ej: encefalopatía hipóxico isquémica), lo cual puede llevar a confusión al momento de localizar. En términos generales la causa de origen central es más frecuente que la periférica, con cifras que varía en los diferentes estudios, con porcentajes de 60-80% versus 15-30%, respectivamente6,11,14.
Los términos hipotonía esencial e hipotonía congénita benigna han perdido su utilidad dado avances en las técnicas diagnósticas. La hipotonía esencial incluía pacientes hipotónicos sin debilidad asociada, en quienes no se encontraba una causa específica, pero que actualmente sabemos tienen diagnósticos reconocidos (ej: Sindrome de Prader Willi) y por otro lado, la hipotonía congénita benigna incluía niños hipotónicos, con y sin debilidad asociada, en quienes tampoco se encontraba causa subyacente, con evolución menos “benigna” a la esperada y que actualmente se sabe son muchas son formas de miopatías congénitas, reconocidas con nuevas técnicas biopsia muscular2,3.
Sindromes hipotónicos centralesUna de las características altamente sugerentes de sindrome hipotónico central, es la presencia de hipotonía en ausencia de debilidad, es decir, logran vencer gravedad y moverse espontáneamente o frente a estímulos táctiles. Generalmente se presentan en el contexto de un retraso global del desarrollo psicomotor. Al examen pueden verse signos de compromiso de primera motoneurona, tales como hiperreflexia, clonus y signo de Babinski. Son orientadores la presencia de compromiso de conciencia, contacto pobre, déficit sensoriales (ej: visual, auditivo), convulsiones, movimientos anormales, reflejos primitivos anormalmente persistentes, rasgos dismórficos, microcefalia y anomalías congénitas mayores, en caso de estar en contexto de un sindrome genético3,12. Ver tabla 2.
Características distintivas de sindromes hipotónicos centrales y periféricos
| Características | Hipotonía Central | Hipotonía Periférica |
|---|---|---|
| Debilidad | Ausente | Presente |
| Reflejos osteotendíneos | Aumentados | Disminuidos o ausentes* |
| Signo Babinski | Presente | Ausente |
| Fasciculaciones | Ausentes | Pueden estar presentes |
| Atrofia | Ausente | Presente |
| Reflejos primitivos | Aumentados (persistentes) | Disminuidos |
| Déficit sensoriales (auditivo/visual) | Pueden estar presentes | Ausente** |
| Encefalopatía, convulsiones, movimientos anormales | Pueden estar presentes | Ausente** |
| Dismorfias | Pueden estar presentes | Raras |
| Microcefalia | Puede estar presente | Rara** |
| Retraso del desarrollo psicomotor | Presente | Presente (retraso motor) |
| CK total | Normal*** | Elevada (ó normal) |
Dentro de las causas de síndromes hipotónicos centrales, la encefalopatía hipóxico isquémica llega hasta un 50% del total, seguida de las enfermedades genéticas con un 15% y malformaciones del sistema nervioso central con un 12%4. Algunas cromosomopatías también se manifiestan con hipotonía de origen central, dentro de las cuales las más prevalentes son: Sindrome de Down, Sindrome Prader Willi y Sindrome Williams. En la tabla 3 se detallan principales características de las condiciones genéticas asociadas a hipotonía.
Características principales de causas frecuentes de hipotonía central
| Diagnóstico | Clínica |
|---|---|
| Trisomía 21 | Perfil facial plano, fisuras palpebrales oblicuas, epicanto, pliegue palmar transversal, clinodactilia del quinto dedo, cardiopatía congénita. |
| Sindrome Prader-Willi | Retraso global del desarrollo, características faciales distintivas (estrechamiento bitemporal, ojos almendrados, estrabismo, labio superior delgado), baja estatura, hipoplasia genital, retraso crecimiento en periodo lactante, luego en infancia hiperfagia y obesidad. |
| Sindrome de Williams | Rasgos faciales distintivos como el hipertelorismo, filtrum largo, boca ancha, micrognatia, estenosis aórtica. |
| Acondroplasia | Prominencia frontal, acortamiento rizomiélico. |
| Trisomía 18 | Restricción del crecimiento intrauterino, occipucio prominente y estrechamiento bitemporal, paladar ojival, micrognatia, uñas hipoplásicas, pie de balancín. |
| Trisomía 13 | Talla baja, holoprosencefalia, labio leporino/paladar hendido, anomalías congénitas múltiples. |
| Sindrome de Angelman | Retraso global del desarrollo, microcefalia adquirida, convulsiones, prognatismo, hipopigmentación cutánea. |
| Espectro MECP2 | Microcefalia adquirida, convulsiones, estereotipias, regresión del desarrollo, predominio femenino. |
| Enfermedades peroxisomales | Disfunción hepática, convulsiones, cataratas, atrofia retiniana, pérdida de la audición, condrodisplasia punctata, fontanela anterior amplia, perfil facial plano. |
(REF. 12).
Una de las principales características de los síndromes hipotónicos periféricos, es la presencia de hipotonía asociada a debilidad y/o contracturas. De hecho, la sensibilidad y especificidad de la ausencia o reducción severa de los movimientos antigravitatorios, indicadores de falta de fuerza muscular, se han estimado entre un 97.4 y 75%, respectivamente, para el diagnóstico de una enfermedad neuromuscular8.
Otros síntomas y signos asociados a hipotonía que orientan a hipotonías de origen periférico son: la presencia de debilidad facial asociada a dismorfismo facial (fascie alargada, paladar ojival, dolicocefalia), lo cual orienta a la presencia de algunas miopatías congénitas específicas (ej: Miopatía nemalínica, miopatía centronuclear, distrofia miotónica tipo 1) y la presencia de ptosis y/u oftalmoplejia (ej: Miopatías centronuclear, multiminicore, Sindromes miasténicos congénitos, enfermedad mitocondrial). La presencia de fasciculaciones sugiere compromiso denervatorio, por compromiso de la motoneurona del asta anterior, altamente sugerente de atrofia muscular espinal (AME). La presencia de contracturas, en especial de artrogriposis, es un indicador de falta de movilidad en el desarrollo fetal temprano (hipo ó akinesia), la cual puede asociarse también a miopatías o neuropatías específicas. El hallazgo de luxación de cadera, especialmente recidivante, pie cavo, escoliosis progresiva y otras deformidades ortopédicas, deben generar la sospecha de una enfermedad neuromuscular de base.
Los pacientes con enfermedades neuromusculares pueden presentar insuficiencia respiratoria secundaria a debilidad de músculos intercostales con indemnidad diafragmática (respiración paradójica, tos débil). Pueden también presentar dificultades en la succión y/o deglución, secundarias a debilidad en la musculatura facial y bulbar, por lo cual pueden presentar dificultad en la eliminación de secreciones. Si bien tanto la debilidad bulbar como el compromiso respiratorio, pueden verse también en síndromes hipotónicos de origen central, una manifestación severa y persistente (mayor a 5 días) obligan a descartar una enfermedad neuromuscular de base, siendo hallazgos frecuentemente encontrados en la miopatía congénita nemalínica severa neonatal, miopatía congénita centronuclear (MTM1, miotubularina), RYR1 severo, distrofia miotónica tipo 1, síndromes miasténicos congénitos, atrofia espinal y enfermedad de Pompe12,15.
Las causas principales de hipotonía periférica clásicamente se han dividido según localización anatómica del compromiso, siendo las más frecuentes la atrofia muscular espinal, distrofia miotónica y miopatías congénitas12. En la tabla 4 se resumen las principales causas de hipotonía periférica hereditaria, con sus características principales.
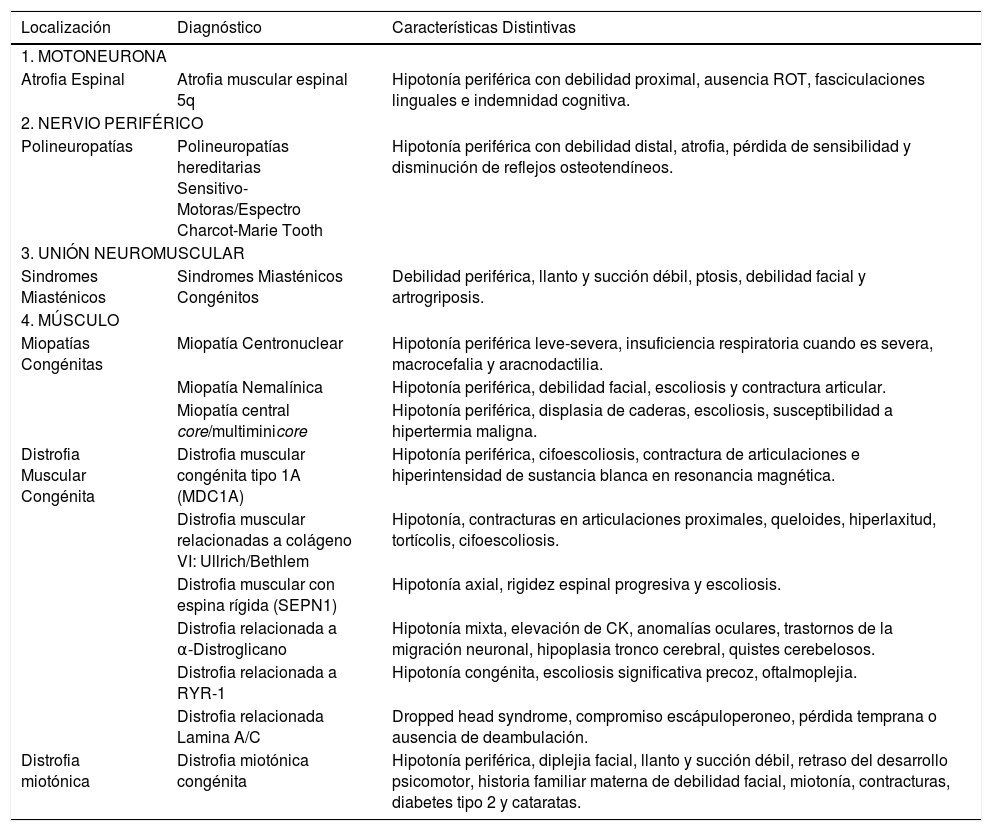
Características distintivas de principales causas de sindrome hipotónico periférico según localización
| Localización | Diagnóstico | Características Distintivas |
|---|---|---|
| 1. MOTONEURONA | ||
| Atrofia Espinal | Atrofia muscular espinal 5q | Hipotonía periférica con debilidad proximal, ausencia ROT, fasciculaciones linguales e indemnidad cognitiva. |
| 2. NERVIO PERIFÉRICO | ||
| Polineuropatías | Polineuropatías hereditarias Sensitivo-Motoras/Espectro Charcot-Marie Tooth | Hipotonía periférica con debilidad distal, atrofia, pérdida de sensibilidad y disminución de reflejos osteotendíneos. |
| 3. UNIÓN NEUROMUSCULAR | ||
| Sindromes Miasténicos | Sindromes Miasténicos Congénitos | Debilidad periférica, llanto y succión débil, ptosis, debilidad facial y artrogriposis. |
| 4. MÚSCULO | ||
| Miopatías Congénitas | Miopatía Centronuclear | Hipotonía periférica leve-severa, insuficiencia respiratoria cuando es severa, macrocefalia y aracnodactilia. |
| Miopatía Nemalínica | Hipotonía periférica, debilidad facial, escoliosis y contractura articular. | |
| Miopatía central core/multiminicore | Hipotonía periférica, displasia de caderas, escoliosis, susceptibilidad a hipertermia maligna. | |
| Distrofia Muscular Congénita | Distrofia muscular congénita tipo 1A (MDC1A) | Hipotonía periférica, cifoescoliosis, contractura de articulaciones e hiperintensidad de sustancia blanca en resonancia magnética. |
| Distrofia muscular relacionadas a colágeno VI: Ullrich/Bethlem | Hipotonía, contracturas en articulaciones proximales, queloides, hiperlaxitud, tortícolis, cifoescoliosis. | |
| Distrofia muscular con espina rígida (SEPN1) | Hipotonía axial, rigidez espinal progresiva y escoliosis. | |
| Distrofia relacionada a α-Distroglicano | Hipotonía mixta, elevación de CK, anomalías oculares, trastornos de la migración neuronal, hipoplasia tronco cerebral, quistes cerebelosos. | |
| Distrofia relacionada a RYR-1 | Hipotonía congénita, escoliosis significativa precoz, oftalmoplejia. | |
| Distrofia relacionada Lamina A/C | Dropped head syndrome, compromiso escápuloperoneo, pérdida temprana o ausencia de deambulación. | |
| Distrofia miotónica | Distrofia miotónica congénita | Hipotonía periférica, diplejia facial, llanto y succión débil, retraso del desarrollo psicomotor, historia familiar materna de debilidad facial, miotonía, contracturas, diabetes tipo 2 y cataratas. |
La atrofia muscular espinal (AME) es una afección autosómica recesiva caracterizada por debilidad muscular progresiva secundaria a degeneración y pérdida de motoneuronas en el asta anterior de la médula espinal y tronco cerebral. Es una de las causas más frecuentes de hipotonía periférica, con una incidencia de 4-10 por 10000016. Existen 4 tipos según edad de inicio y severidad (Tipo 0 ó 1A de inicio pre/perinatal, al tipo 4 adulto), ninguno de los cuales presenta compromiso cognitivo. Los pacientes AME tipo 1 se presentan en los primeros 6 meses de vida con hipotonía periférica, retraso en el desarrollo motor, mayoría con fasciculaciones linguales y ausencia de reflejos osteotendinosos. Presentan respiración paradójica dada debilidad en la musculatura intercostal, además de dificultades en la deglución que, en caso de no tratarse, afectan el desarrollo pondoestatural. Estos pacientes nunca logran sentarse y la historia natural ha mostrado que la mayoría fallece antes de los 2 años, principalmente por el compromiso respiratorio, de no mediarse alguna intervención de ventilación asistida y/o trasqueostomía, que prolongue su sobrevida. Los pacientes con AME tipo 2 logran sentarse, sin embargo no logran marcha independiente, con expectativas de vida hasta 30-50 años. Los pacientes AME tipo 3 logran cierta marcha independiente, presentando caídas y dificultad en subir escaleras alrededor de los 2 años, teniendo una expectativa de vida normal. En cambio los pacientes AME tipo 4 inician síntomas en la segunda o tercera década de vida, con evolución mucho más estable y suelen deambular sin mayores complicaciones durante toda la vida. El diagnóstico en todos los tipos se confirma con estudio genético del gen de sobrevida de la motoneurona (SMN1), que en un 95-98% de los casos muestra deleción homocigota del exón 717–19. En el último tiempo, dados los avances en la investigación de nuevas terapias, tales como oligonucleótido antisentido, recientemente aprobada y la terapia génica con resultados prometedores, han cambiado el paradigma de enfermedad intratable.
Distrofia miotónica congénitaLa distrofia miotónica tipo 1 (DM1) es un trastorno multisistémico autosómico dominante, que causa debilidad de la musculatura esquelética y lisa, involucrando ojo, corazón y otros órganos. Se clasifica en 3 subtipos: leve, clásica y congénita, con una incidencia de 1:2000020. La DM1 de presentación congénita debe estar en el diagnóstico diferencial de todo neonato con debilidad muscular severa y compromiso respiratorio. En la historia pueden encontrarse antecedentes de disminución de movimientos fetales y/o polihidroamnios. Presentan debilidad facial (diplejia facial), labio superior invertido y paladar ojival. El llanto débil y pobre succión ocurren hasta en un 75% de los casos, y la falla respiratoria puede ser fatal21. Aquellos que sobreviven, generalmente muestran mejorías en su hipotonía, sin embargo la debilidad facial permanece o se acentúa en el tiempo y hasta un 50% presenta deficiencia intelectual de severidad variable22. En la electromiografía ayuda al diagnóstico dado la presencia de descargas miotónicas. Deben evaluarse periódicamente con ECG o Holter, dado la potencialidad de arritmias por defectos en la conducción que son asintomáticos. La DM1 se debe a expansión del nucleótido CTG del gen DMPK, las cuales se producen más frecuentemente por transmisión materna. Debido a esto, ante la sospecha de una DM1, debe evaluarse siempre a la madre, buscando debilidad facial y miotonía (dificultad en la relajación muscular) evaluadas en la prehensión palmar o ante la percusión muscular, junto con los hallazgos en la electromiografía.
Miopatías congénitasLas miopatías congénitas (MC) son un grupo heterogéneo de enfermedades hereditarias del músculo, que frecuentemente se manifiestan desde el nacimiento con anomalías estructurales características de las fibras musculares. Se presentan generalmente desde el período de recién nacido o lactante, con hipotonía periférica, debilidad muscular generalmente proximal (debilidad de cintura escapular y pelviana) y retraso en la adquisición de hitos motores, de severidad variable, siendo mayoría de evolución benigna no progresiva. Pueden presentar debilidad facial y de la musculatura oral, con paladar ojival y dificultades en la alimentación, además de debilidad en la musculatura respiratoria que puede llevar a necesidad de apoyo ventilatorio nocturno. La CK puede estar normal o discretamente elevada y el estudio electrofisiológico puede mostrar potenciales motores polifásicos y de baja amplitud. Históricamente, la biopsia muscular junto con el estudio de inmunohistoquímica y microscopía electrónica, han clasificado a las miopatías en la base de los hallazgos patológicos más característicos, dividiéndolas en miopatía por desproporción de fibras, miopatía nemalinica, central core, multiminicore, centronuclear y miotubular. Ver tabla 4. En los últimos 20 años se han ido reconociendo en forma creciente los genes responsables para cada una, existiendo una gran heterogenicidad genética y superposición clínica, de hecho cada miopatía congénita puede ser causada por mutaciones en más de un gen y por otro lado, mutaciones en un mismo gen pueden producir diferentes hallazgos patológicos en la biopsia muscular15.
Distrofia Muscular CongénitaLas distrofias musculares congénitas (DMC) son un grupo heterogéneo de enfermedades musculares hereditarias de inicio precoz, en quienes la biopsia muscular muestra un patrón distrófico, sin evidencia histológica de otra enfermedad neuromuscular23. Las características clínicas varían desde afecciones severas y frecuentemente fatales, hasta leves compatibles con una vida adulta24. En la medida que se han ido identificando y conociendo nuevos defectos genéticos y bioquímicos, la clasificación clínica ha ido cambiando, incorporando estos a la nomenclatura referidos como “distrofia relacionada” al defecto genético o proteína afectada (tabla 4). En términos generales los pacientes con DMC presentan hipotonía periférica con debilidad asociada y CK generalmente elevada, sin embargo la presencia de signos de compromiso de SNC (ej: retraso global desarrollo psicomotor, crisis convulsivas, espasticidad, signos piramidales y malformaciones del desarrollo SNC), pueden verse en pacientes con DMC del tipo alfaDG-RD, siendo diagnóstico diferencial de síndromes hipotónicos centrales y mixtos. La presencia de hiperlaxitud especialmente distal, asociada a contracturas proximales y axiales y luxación congénita de cadera, es altamente sugerente de la DMC secundaria a afección del colágeno VI (COLVI-RD) y por otro lado, la debilidad axial con “dropped head”, altamente sugerente de laminopatía (LMNA) o afección de selenoproteína (SEPN1). En cambio, la presencia de debilidad facial predominante, oftalmoplejia precoz, falla respiratoria temprana y cardiomiopatía congénita, no son hallazgos frecuentes en DMC, debiendo considerarse otras alternativas diagnósticas en primer lugar (miopatías congénitas, DM1, entre otros).
Sindromes miasténicos congénitosLos síndromes miasténicos congénitos son un grupo de enfermedades caracterizadas por debilidad y fatigabilidad de la musculatura ocular, bulbar y de extremidades, con indemnidad de musculatura cardíaca y lisa. Se presentan desde el nacimiento hasta la niñez, con dificultades en la alimentación, succión débil, llanto débil, ptosis, debilidad facial y artrogriposis. Pueden presentar insuficiencia respiratoria, la cual puede se de curso rápido llevando a apnea y cianosis. En lactantes puede verse fatigabilidad, con debilidad fluctuante que empeora a lo largo del día, y ptosis o debilidad ocular intermitente. El diagnóstico se puede realizar a través del estudio electrofisiológico, con un test de estimulación repetitiva (TER) o fibra única alterada, que pone de manifiesto la alteración en la unión neuromuscular, lo cual en conjunto a la presencia de anticuerpos contra el receptor de acetilcolina y el estudio genético, confirman el diagnóstico3.
Polineuropatías Charcot-Marie ToothLas polineuropatías hereditarias sensitivo motoras o Polineuropatías Charcot-Marie Tooth (CMT) son un grupo heterogéneo de enfermedades de los nervios periféricos, siendo la más común, la asociada a desmielinización con debilidad distal de extremidades, atrofia y déficit sensitivo, disminución en los ROTs y velocidad de conducción nerviosa reducida en el estudio electrofisiológico. El Sindrome de Dejerine-Sottas, o CMT tipo III es uno de los subtipos desmielinizantes severos que puede presentarse desde la infancia, con velocidades de conducción severamente reducidas (<10m/seg), de herencia autosómica recesiva, por mutaciones en los genes PMP22, MPZ o ERG2.
ENFOQUE DIAGNÓSTICOExisten múltiples enfoques y algoritmos descritos de aproximación ante un lactante o neonato con hipotonía1,3,12,25. Como se mencionó anteriormente, lo más importante es intentar localizar si se trata de un sindrome hipotónico central, periférico o mixto, a través de una historia exhaustiva y examen físico detallado. En caso de un sindrome hipotónico central, lo primero es identificar diagnósticos potencialmente mortales y causas tratables (ej: sépticas, metabólicas, crisis epilépticas, entre otros), para luego considerar las etiologías más comunes, algunas de las cuales se reconocen en la primera evaluación (ej: Sd Down). Los exámenes que más tienen rendimiento diagnóstico en caso de no tener sospecha clínica, son los estudios de imágenes (TAC, RM Cerebral) que pueden descartar anomalías estructurales del sistema nervioso, secuela de asfixia, entre otras. La evaluación genética puede definir si está en el contexto de un sindrome genético reconocible, solicitar estudio genético específico, además de descartar posibles malformaciones mayores asociadas, a través de la evaluación de la visión, audición, ecocardiograma y eco abdominal3,5,11,12. Estudios realizados a neonatos con hipotonía (central y periférica), han mostrado que el estudio dirigido con next-generation sequencing (NGS), han aumentado la tasa diagnóstica en un 22%, después de realizados métodos genéticos tradicionales (ej. Cariotipo molecular, MLPA, metilación, PCR, entre otros)14.
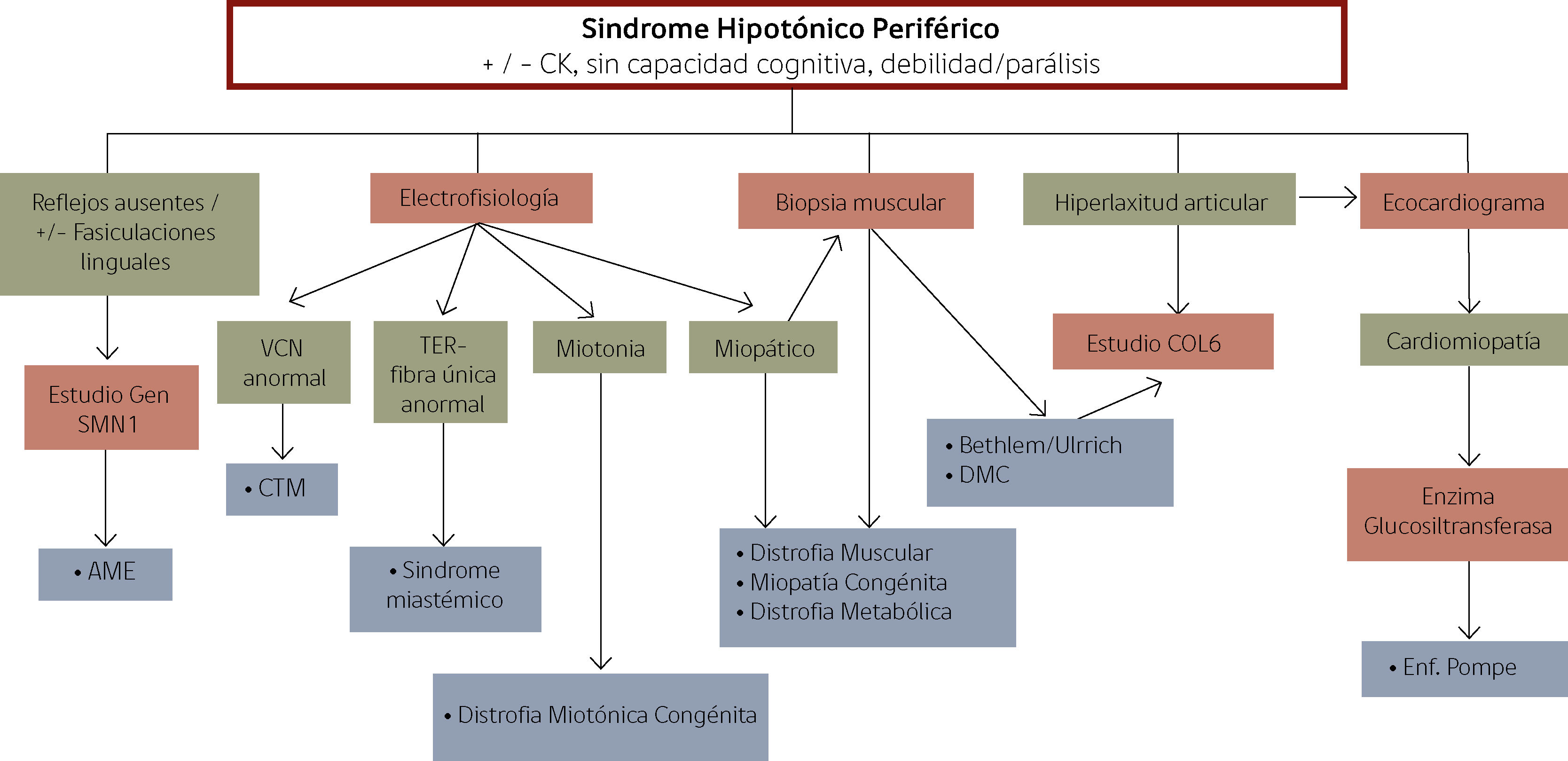
Frente a la sospecha de un sindrome hipotónico periférico, los estudios diagnósticos deben inclinarse en esa dirección (CK total, electrofisiología, biopsia muscular), según la sospecha diagnóstica (ej: estudio gen SMN1 frente a sospecha de atrofia espinal). Un nivel de CK elevada orienta a causa periférica (ej: distrofia muscular congénita), sin embargo debe interpretarse con cautela en recién nacidos, dado que puede elevarse en forma transitoria en primeros días post-parto. Por otro lado, niveles normales de CK no descartan compromiso periférico, estando normales por ejemplo en varias miopatías congénitas y distrofia miotónica12. El estudio electrofisiológico puede ser de gran utilidad en el diagnóstico de neuropatías periféricas, distrofia miotónica y miastenias congénitas y la biopsia muscular puede orientar en el diagnóstico de miopatías congénitas, distrofias musculares, junto con el estudio genético específico y resonancia muscular12. La figura 2 presenta un flujograma más detallado de los principales aspectos a considerar al enfrentarnos a un neonato con sospecha de sindrome hipotónico periférico.

Enfrentamiento inicial de lactante con Sindrome Hipotónico Periférico
CK: Creatinquinasa, SMN1: Proteina para la supervivencia de la neurona, AME: Atrofia Muscular Espinal, VCN: Velocidad de conducción nerviosa, TER: Test de estimulación repetitiva, DMC: Distrofia Muscular Congénita y COL6: Colágeno tipo VI.
Adaptado de Lisi E et al., 3.
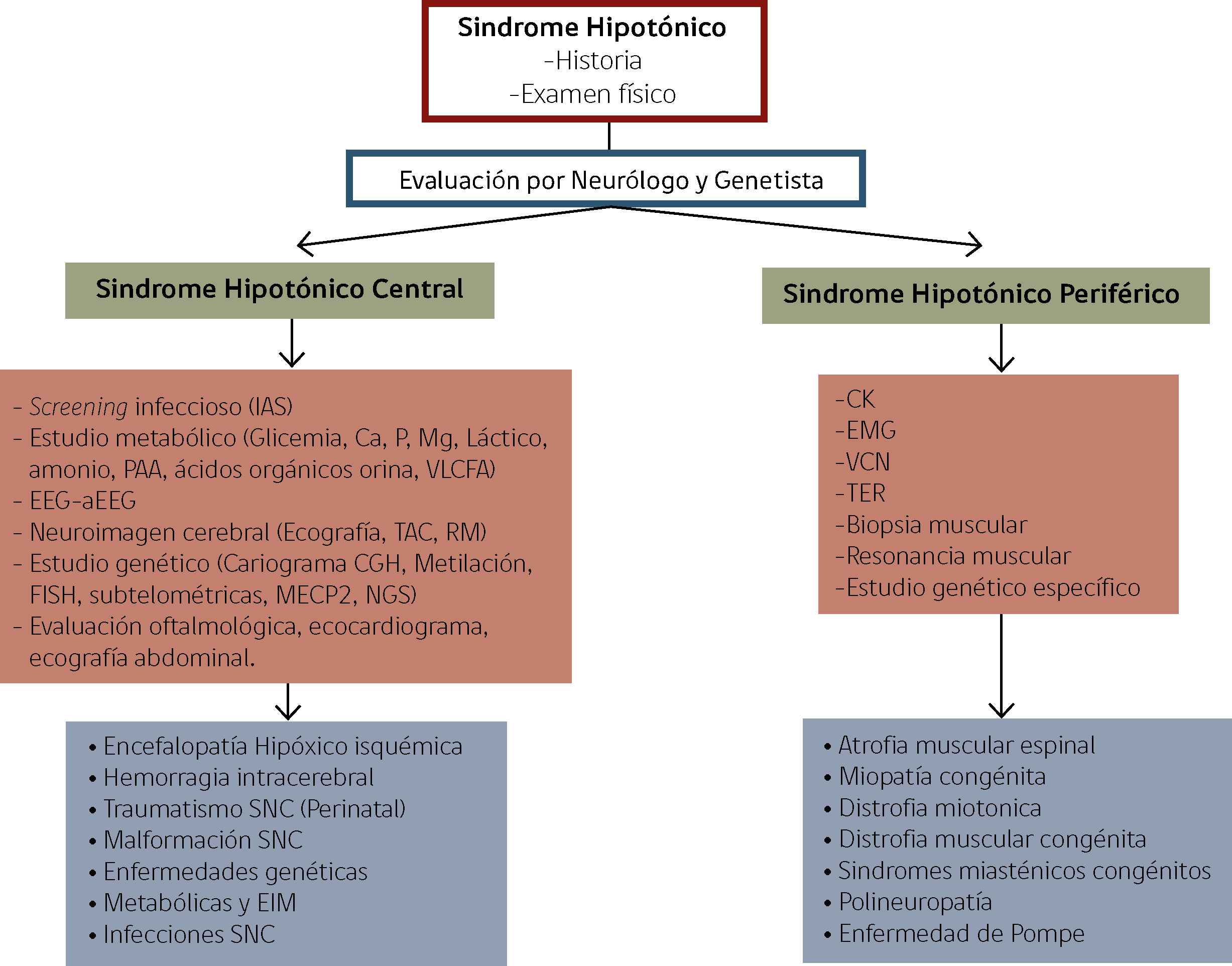
La figura 3 presenta un flujograma de enfrentamiento del lactante con sindrome hipotónico central ó periférico. Los exámenes deben ser solicitados gradualmente, considerando contexto clínico y sospecha diagnóstica.
.")
Enfrentamiento de Lactante con Sindrome Hipotónico
Ca: calcio, P. Fósforo, Mg: Magnesio, PAA: Perfil de aminoácidos y acilcarnitinas, VLCFA: Ácidos grasos de cadena muy larga, EEG: electroencefalograma, aEEG: electro encefalograma de amplitud integrada, TAC: Tomografía axial computarizada, RM: Resonancia magnética, CGH: Hibridación genómica comparativa, FISH: Hibridación fluorescente in situ, MECP2: Metil CpG binding protein 2, NGS: Next generation sequencing, CK: Creatinquinasa, EMG: Electromiografía, VCN: Velocidad de conducción nerviosa, TER: Test de estimulación repetitiva, SNC: Sistema nerviosos Central, EIM: Errores innatos del metabolismo y CMT: Charcot Marie Tooth.
Adaptado de Prasad A, Prasad C & De Vivo M, et al. (9,25).
La hipotonía es un signo inespecífico de un grupo amplio de patologías, donde la historia clínica y examen físico siguen siendo cruciales en la orientación diagnóstica. Si bien la mayoría de los casos son hipotonías centrales, las causas de origen periférico o neuromuscular han ido cobrando importancia dadas nuevas técnicas diagnósticas. La importancia en llegar a un diagnóstico específico tiene implicancias en el pronóstico de los pacientes, en la prevención y manejo de complicaciones asociadas a cada patología, además de realizar consejo y asesoría genética. Por otro lado, llegar un diagnóstico ha permitido mejorar la sobrevida y calidad de vida de estos pacientes, asociado en especial a los grandes avances en los cuidados y en la investigación de terapias del último tiempo.
Declaración de interésLos autores declaran no tener conflictos de interés en relación al presente artículo.