







La Poliposis Adenomatosa Familiar (PAF) y el Síndrome de Lynch (SL) son los dos síndromes hereditarios más importantes que predisponen al cáncer colorrectal (CCR). La PAF surge de mutaciones en el gen APC, y causa CCR en todos los portadores después de los 40 años. La enfermedad se expresa desde la adolescencia y puede ser tratado con cirugía profiláctica. Como otras causas de muerte en pacientes con PAF se observan el cáncer periampular y los tumores desmoides.
En cambio, el SL es causado por mutaciones en los genes de reparación del DNA, más frecuentemente MLH 1, MSH 2 y MSH 6. El diagnóstico clínico se basa en los criterios de Amsterdam. La penetrancia en portadores para CCR es incompleta, por lo que la colectomía profiláctica es controvertida. El SL se asocia con alta frecuencia a otros tipos de tumores, como el cáncer de endometrio, cáncer de ovario y cáncer de urotelio entre otros.
En ambos síndromes, el estudio genético de los pacientes permite en altos porcentajes identificar las mutaciones causantes. En base a estos resultados, se pueden identificar los familiares portadores de mutaciones, y ofrecerles los esquemas de vigilancia y eventuales tratamientos profilácticos correspondientes a su enfermedad. Para el manejo integral de estas familias es esencial contar con un equipo multidisciplinario, para poder ofrecer consejería genética, estudio genético, educación a las familias y tratamiento respectivo en forma adecuada.
Familial Adenomatous Polyposis (FAP) and Lynch Syndrome (LS) are the most important hereditary syndromes that predispose to colorectal cancer (CRC).
FAP is caused by mutations in the APC gene, and leads to CRC in all mutations carriers beyond age 40. Disease expression starts during adolescence, and can be treated by prophylactic surgery. Periampullar cancer and desmoid tumors account for disease-related deaths after CRC.
On the other hand, LS is caused by mutations of mismatch repair genes such as MLH1, MSH2 and MSH. The clinical diagnose is based on Amsterdam criteria. Penetrance for CRC is incomplete, wherefore prophylactic colectomy is controversial. LS is frequently associated with other cancers such as endometrium, ovaries and urothelium.
In both syndromes, the genetic study of the patients offers with a high frequency the identification of the underlying mutations. Based on these results, mutations carriers can be identified among the family members, which offers them the opportunity for surveillance and prophylactic treatment according to their disease pattern. In order to guarantee an integrated health care management for these families, it is essential to count on a multidisciplinary team offering genetic counselling, molecular genetic studies, education and respective treatment in an adequate fashion.
Los síndromes hereditarios que predisponen al desarrollo del cáncer colorrectal se pueden dividir en dos grandes grupos: Los síndromes polipósicos, y dentro de ellos la Poliposis Adenomatosa, Familiar (PAF) como la enfermedad más importante, y el cáncer colorrectal hereditario no polipósico (CCHNP), cuyo representante principal es el Síndrome de Lynch. El objetivo de este artículo es de acercarle al lector a estos dos síndromes más importantes, su presentación clínica, información genética y opciones de prevención y tratamiento.
POLIPOSIS ADENOMATOSA FAMILIAR (PAF)La PAF es una enfermedad autosómica dominante causada por mutaciones en el gen APC ubicado en el cromosoma 5q. Los portadores desarrollan múltiples pólipos adenomatosos en colon y recto (>100), en algunas formas incluso sobre 1000, distribuidos en todo el colon y recto con predominio en el colon sigmoides. La incidencia se describe entre 10 y 14 en 100000 personas y la penetrancia, es decir la tasa de portadores que desarrollan cáncer de colon y recto (CCR) es del 100%, es decir cada portador desarrollará CCR hasta la edad de 45 años 1,2.
En algunos pacientes, los pólipos se desarrollan ya en la primera década de vida, pero en la mayoría de los casos se diagnostica en la pubertad (entre los 12 y los 16 años). Hay que considerar, que casi todos los portadores a la edad de 35 años tendrán adenomas, y la progresión hacia el adenocarcinoma es inevitable 3. La edad promedio de diagnóstico del cáncer es de 39 años, con una frecuencia global de 7% a los 21 años y 95% a los 50 años 4.
Los fenotipos de los pacientes son variables, al igual que las manifestaciones extracolónicas. Durante muchos años, la causa de muerte principal en pacientes con PAF fue el CCR. Con el aumento del conocimiento sobre la enfermedad, mejores alternativas diagnósticas y la introducción de la colectomía profiláctica, las manifestaciones extracolónicas malignas han cobrado cada vez más importancia como causa de muerte. Estás incluyen el cáncer periampular, cáncer gástrico, tumores desmoides, carcinoma suprarrenal, colangiocarcinoma, hepatoblastoma, glioblastoma entre otros (Tabla 1). También existen manifestaciones extracolónicas benignas, como adenomas gástricos y duodenales, adenomas suprarrenales, osteomas, dentición supranumeraria, quistes epidermoides y la hipertrofia congénita del epitelio pigmentado de la retina (CHRPE). Se ha observado que hay cierta correlación entre diferentes tipos de mutaciones y el desarrollo de manifestaciones extracolónicas 5, por lo que el estudio genético ayuda a definir subgrupos de pacientes con PAF con alto riesgo de tumores extracolónicos y orienta tanto en la conducta terapéutica en estos pacientes como en la selección de exámenes de vigilancia específica.
MANIFESTACIONES EXTRAINTESTINALES EN LA POLIPOSIS ADENOMATOSA FAMILIAR
| Lesiones benignas | Lesiones malignas | ||
|---|---|---|---|
| Hipertrofia congénita del epitelio pigmentario de la retina | 70-80% | Cáncer tiroides | 2-3% |
| Quistes epidermoides | 50% | Tumores cerebrales | <1% |
| Osteoma | 50-90% | Hepablastoma | 1% |
| Tumores desmoides | 10-15% | ||
| Dientes supernumerarios | 11-27% | ||
| Adenomas suprarrenales | 7-13% | ||
Vasen. GUT 2008.
Las manifestaciones extracolónicas del PAF más frecuentes son:
Pólipos del tracto digestivo superior, presentándose adenomas gástricos y/o duodenales en casi el 90% de los pacientes a una edad media de 38 años 1,2. Comparado con la población general, los portadores de PAF tienen un mayor riesgo de tener pólipos de glándulas fúndicas (25-60% versus 1-2%) 6.
Los adenomas duodenales, de la papila de Vater y periampulares se observan en el 58% a 74% de los portadores de PAF y el cáncer periampular es la segunda causa de muerte por cánceres extracolónicos en estos pacientes después de los tumores desmoides.
Pólipos gastroduodenales se clasifican según Spigelman (Tabla 2).
CLASIFICACIÓN DE SPIGELMAN
| Criterio | 1 punto | 2 puntos | 3 puntos |
|---|---|---|---|
| Número de pólipos | 1-4 | 5-20 | >20 |
| Tamaño de pólipos (mm) | 1-4 | 5-10 | >10 |
| Histología | Tubular | Tubulo-velloso | Velloso |
| Displasia | Bajo grado | Bajo grado | Alto grado |
Estadio 0:0 puntos, estadio I: 1-4 puntos, estadio II: 5-6 puntos, Estadio III: 7-8 puntos, estadio IV: 9-12 puntos.
Spigelman. Lancet 1989.
Desarrollo de un cáncer periampular se observa en el 1.6% de los pacientes con Spigelman IV en la endoscopia digestiva alta.
En consecuencia, hay que realizar esta endoscopia en todos los pacientes con PAF mínimo cada 3 años (según hallazgos, ver tabla), dado que el riesgo de cáncer periampular en portadores PAF es 100 a 300 veces mayor que en la población general 7,8.
Tumores del sistema nervioso central (SNC) se describen como Síndrome de Turcot. Si se asocian con adenomas periampulares, CHRPE, osteomas mandibulares, odontomas, tumores desmoides, tumores tiroideos y/o de la vía biliar, se denomina Síndrome de Gardner 5.
Tumores desmoides ocurren en el 10 a 30% de los casos, son de alta morbilidad (sobre todo los desmoides mesentéricos) y el riesgo de desarrollarlos aumenta con una historia familiar positiva para desmoides, mutación del codón 1309, el género femenino y cirugía abdominal previa 9,10.
Existe una variación fenotípica de la PAF clásica conocida como PAF atenuada (PAFA), donde se observan menos de 100 pólipos en colon y recto (entre 10 y 100). El inicio de la expresión es más tardío alrededor de los 50 años de vida, y los pólipos se concentran más en el colon proximal. Las manifestaciones extracolónicas son parecidas a las de la PAF clásica, y el riesgo cumulativo de CCR es más bajo (aprox. 65%). Esta variante no se debe a mutaciones del gen APC, sino a mutaciones en el gen MYH en el cromosoma 1p (MUTYH), que se hereda en forma recesiva 11.
Diagnóstico clínico y molecularEl diagnóstico clínico de la PAF es difícil, dado que la enfermedad en general sobre todo en estadíos iniciales no da síntomas, o síntomas muy inespecíficos. Los pólipos pueden causar desde rectorragia, secreción mucosa con las deposiciones, o anemia hasta obstrucciones intestinales, dependiendo de número, tamaño y ubicación de los pólipos. Otros síntomas inespecíficos son dolor abdominal, cambios en el hábito intestinal y baja de peso. En general, los síntomas se presentan más bien en pacientes con enfermedad avanzada y/o cáncer.
Por eso, es importante realizar exámenes preventivos de vigilancia a todo portador desde los 12-14 años de edad. El examen de elección es la colonoscopia larga, para pesquisar precozmente el inicio de la expresión de la enfermedad 12.
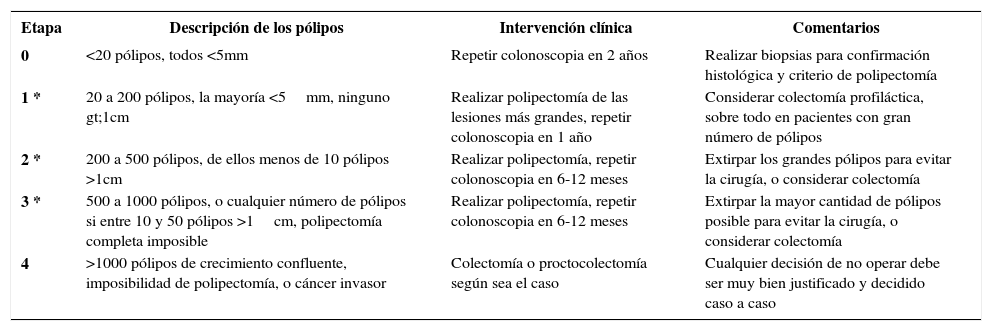
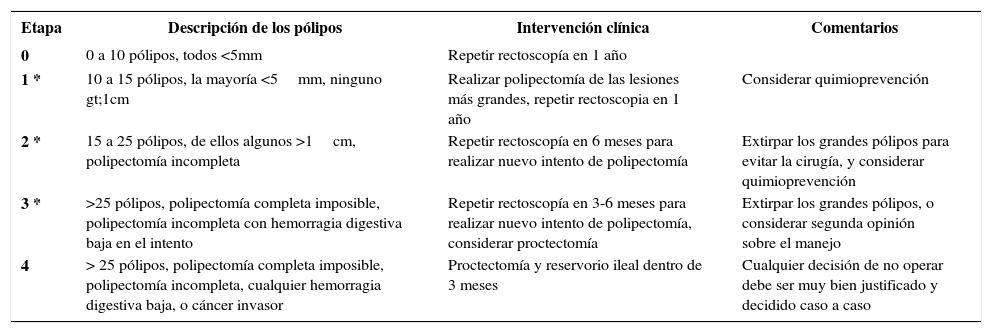
La PAF se clasifica de acuerdo al número de pólipos encontrados en la colonoscopía: PAF clásica en caso de más de 100 pólipos, PAF severa o profusa con más de 1000 pólipos, y atenuada entre 10 y 100 pólipos y edad más avanzada (4ta y 5ta década de vida) 13,14. En 2016, la Sociedad Internacional para Tumores Gastrointestinales Hereditarios (InSiGHT) estableció un sistema de etapificación más preciso para las PAF, con el fin de poder elegir el mejor manejo de estos pacientes según su grado de avance de la enfermedad ya que sea mediante quimioprevención, manejo endoscópico o cirugía profiláctica 15. Esta clasificación considera el número y tamaño de los adenomas, pero no incluye factores como edad del paciente, grado de displasia o manifestaciones extracolónicas. Para el manejo de pacientes no operados, se establecen los criterios de manejo endoscópico versus quirúrgico del colon (InSiGHT polyposis staging system – colon: IPSS-colon) (Tabla 3), y para pacientes ya operados de una colectomía se aplica la clasificación para el manejo del recto remanente (InSiGHT polyposis staging system – rectum: IPSS-rectum) (Tabla 4).
SISTEMA DE ETAPIFICACIÓN DE POLIPOSIS DE COLON INSIGHT
| Etapa | Descripción de los pólipos | Intervención clínica | Comentarios |
|---|---|---|---|
| 0 | <20 pólipos, todos <5mm | Repetir colonoscopia en 2 años | Realizar biopsias para confirmación histológica y criterio de polipectomía |
| 1 * | 20 a 200 pólipos, la mayoría <5mm, ninguno gt;1cm | Realizar polipectomía de las lesiones más grandes, repetir colonoscopia en 1 año | Considerar colectomía profiláctica, sobre todo en pacientes con gran número de pólipos |
| 2 * | 200 a 500 pólipos, de ellos menos de 10 pólipos >1cm | Realizar polipectomía, repetir colonoscopia en 6-12 meses | Extirpar los grandes pólipos para evitar la cirugía, o considerar colectomía |
| 3 * | 500 a 1000 pólipos, o cualquier número de pólipos si entre 10 y 50 pólipos >1cm, polipectomía completa imposible | Realizar polipectomía, repetir colonoscopia en 6-12 meses | Extirpar la mayor cantidad de pólipos posible para evitar la cirugía, o considerar colectomía |
| 4 | >1000 pólipos de crecimiento confluente, imposibilidad de polipectomía, o cáncer invasor | Colectomía o proctocolectomía según sea el caso | Cualquier decisión de no operar debe ser muy bien justificado y decidido caso a caso |
* La presencia de displasia de alto grado justifica clasificación del paciente en etapa 4
Fuente: Lynch PM et al. 4
SISTEMA DE ETAPIFICACIÓN DE POLIPOSIS RECTAL INSIGHT
| Etapa | Descripción de los pólipos | Intervención clínica | Comentarios |
|---|---|---|---|
| 0 | 0 a 10 pólipos, todos <5mm | Repetir rectoscopía en 1 año | |
| 1 * | 10 a 15 pólipos, la mayoría <5mm, ninguno gt;1cm | Realizar polipectomía de las lesiones más grandes, repetir rectoscopia en 1 año | Considerar quimioprevención |
| 2 * | 15 a 25 pólipos, de ellos algunos >1cm, polipectomía incompleta | Repetir rectoscopía en 6 meses para realizar nuevo intento de polipectomía | Extirpar los grandes pólipos para evitar la cirugía, y considerar quimioprevención |
| 3 * | >25 pólipos, polipectomía completa imposible, polipectomía incompleta con hemorragia digestiva baja en el intento | Repetir rectoscopía en 3-6 meses para realizar nuevo intento de polipectomía, considerar proctectomía | Extirpar los grandes pólipos, o considerar segunda opinión sobre el manejo |
| 4 | > 25 pólipos, polipectomía completa imposible, polipectomía incompleta, cualquier hemorragia digestiva baja, o cáncer invasor | Proctectomía y reservorio ileal dentro de 3 meses | Cualquier decisión de no operar debe ser muy bien justificado y decidido caso a caso |
* La presencia de displasia de alto grado justifica clasificación del paciente en etapa 4
Fuente: Lynch PM et al. 4
Como indican las guías y consensos internacionales 16, el estudio genético se realiza generalmente en el marco de una consejería genética en los pacientes afectados por la enfermedad, cuyo diagnóstico fue confirmado clínica y endoscópicamente. Si se identifica la mutación en el individuo afectado, se puede buscar dirigidamente la mutación en sus familiares en riesgo (diagnóstico molecular) y con esta información realizar la vigilancia clínica y endoscópica solamente en los portadores identificados, liberando de esta forma a los familiares sin mutación de la vigilancia de por vida por le hecho que presentan un riesgo para CCR igual a la población general. En caso de que no se logre identificar una mutación en el paciente índice, hay que advertirle al paciente que esto muy probablemente se deba a que no se tiene la capacidad técnica de pesquisar la mutación, por lo que todos los familiares en riesgo tienen que someterse a exámenes de vigilancia en forma periódica según los esquemas acordados en los consensos ya mencionados 16.
Los estudios genéticos tienen que realizarse en torno a un grupo de profesionales experimentados en el manejo de enfermedades hereditarias, ya que se requiere una consejería genética amplia y completa tanto para el paciente como sus familiares, que abarca tanto aspectos médicos, técnicos y éticos, como orientación psicológica y contención emocional 17.
Los estudios genéticos del gen APC pueden tener 3 posibles resultados. En el primer caso, se logra identificar la mutación causante de la enfermedad, lo que permite estudiar a todos los familiares en riesgo, con eso separarlos en portadores y no portadores, y luego realizarles seguimiento y vigilancia a los portadores. Si no se logra identificar una mutación en APC incluso con estudios repetidos, se recomienda realizar análisis del gen MYH para descartar la presencia de una PAFA. En algunos casos, se pueden diagnosticar variantes de significado incierto (VUS, “variants of uncertain significance”), que requieren una consejería individualizada ajustada al caso específico, considerando todos los factores involucrados 1.
Tratamiento farmacológico – QuimioprevenciónNo existe tratamiento farmacológico que sea capaz de eliminar la enfermedad o detener su progreso completamente. Sin embargo, se ha demostrado que la administración de anti inflamatorios inhibidores de COX-2 (específicamente celecoxib), es capaz de disminuir el número de nuevos adenomas y disminuir la velocidad de crecimiento de ellos 18.
En pacientes con una poliposis en estadío inicial, se puede usar este tratamiento como puente para enlentecer el progreso de la enfermedad y postergar la cirugía profiláctica (quimioprevención). Esta estrategia es especialmente útil en pacientes adolescentes, para poder postergar la cirugía profiláctica hasta que avance su desarrollo físico y psicosocial a un nivel compatible con una cirugía mayor. En pacientes operados con preservación de recto, se puede utilizar como tratamiento adyuvante para frenar el progreso de la enfermedad en el recto remanente y de esta manera, hacer la enfermedad más susceptible a un manejo endoscópico eficiente 19.
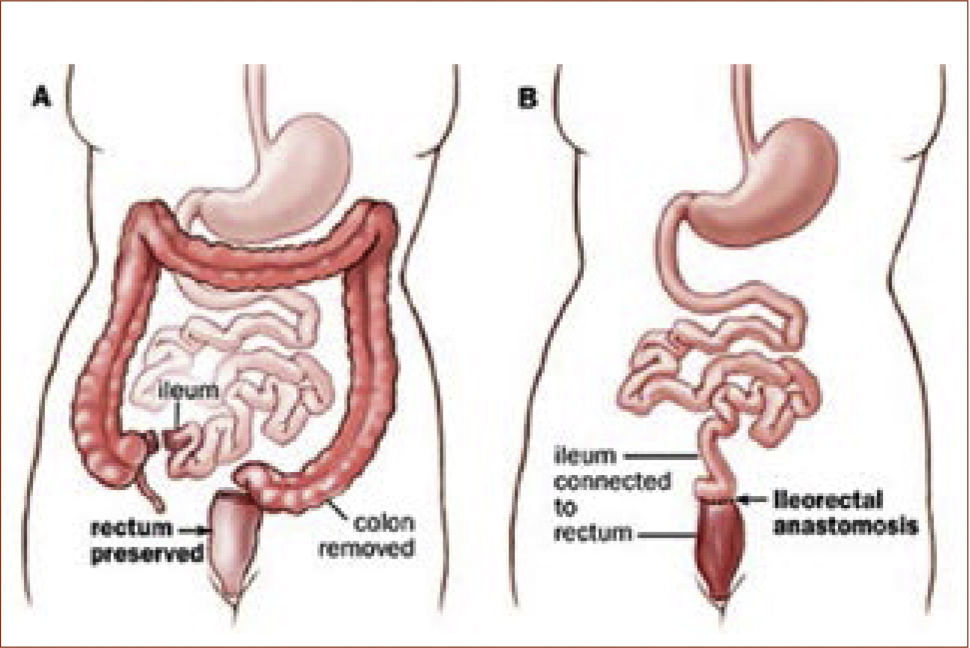
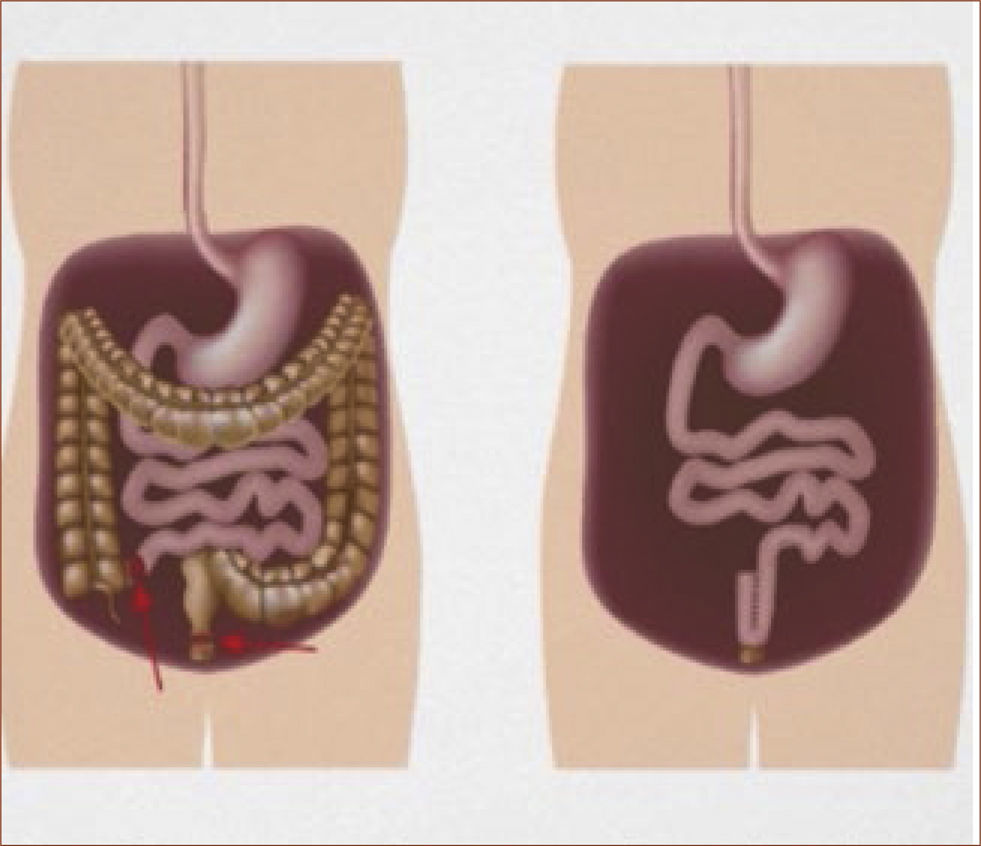
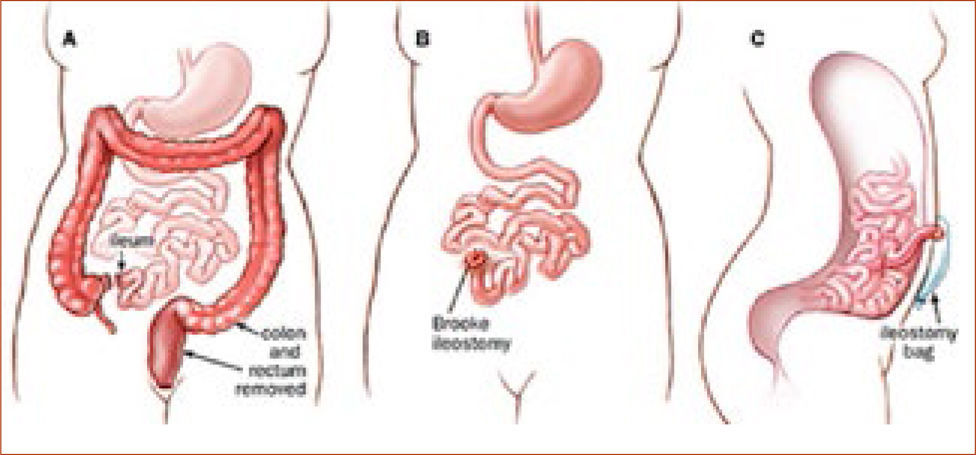
Tratamiento quirúrgicoSegún lo mencionado anteriormente, el tratamiento de la PAF consiste en cirugía profiláctica en pacientes cuando el cuadro clínico lo indica (etapificación IPSS, (Tabla 3). La indicación quirúrgica se basa en el riesgo de desarrollar cáncer. El tipo de operación a realizarse depende del compromiso del paciente, básicamente en el recto (Tabla 4) y una eventual disposición al desarrollo de tumores desmoides (tipo de mutación y/o historia familiar). En el caso de un escaso o moderado compromiso rectal que se puede manejar en forma endoscópica, se recomienda remover el colon completo y realizar una ileo recto anastomosis (ver Figura 1). En el caso de un compromiso severo de recto, displasia de alto grado y/o cáncer de recto, se requiere una resección completa de colon y recto (panproctocolectomia), y una reconstrucción compleja mediante reservorio ileal y anastomosis ileo anal (Figura 2). En algunos casos, el o la paciente puede ser no apto para recibir un reservorio ileal (incontinencia fecal previa, daños esfinteriano, radioterapia, compromiso del esfínter anal por tumor). En estos casos, se procede a realizar la proctocolectomia sin reconstrucción, con una ileostomía terminal definitiva (Figura 3). Cabe destacar, que estas cirugías son de alta complejidad y deben ser realizados solamente por cirujanos coloproctólogos especializados, en el marco de un equipo y una institución con vasta experiencia en este tipo de operaciones.



Es el cáncer hereditario más frecuente estimándose que corresponde al 3-5% del total de CCR. Es un síndrome que se hereda en forma autosómica dominante y es causado por una mutación germinal en uno de los genes que participan en la reparación del ADN (mismatch repair genes). Los portadores de las mutaciones presentan un riesgo muy elevado de CCR, cáncer de endometrio y otros tumores (ovario, uroepitelio, estómago, intestino delgado, vías biliares y páncreas, piel como adenomas sebáceos, carcinomas y queratoacantomas y cerebro).
La primera comunicación científica al respecto fue realizada por Aldred Warthin en al año 1913 en la revista Archives of Internal Medicine. Este patólogo de la Universidad de Michigan publicó el caso de su costurera, deprimida por el temor a fallecer por cáncer a una temprana edad, debido a sus antecedentes familiares. Efectivamente, falleció de cáncer de endometrio. La importancia de esta primera descripción no fue valorada hasta 1966, cuando Henry T. Lynch publica su experiencia sobre dos familias que denominó “Familias N y M” introduciendo la denominación de «síndrome de cáncer familiar» 20.
Características clínicasExisten dos formas de presentación clínica:
Tipo I cuando afecta exclusivamente colon y recto
Tipo II cuando se afectan otros órganos.
La penetrancia (porcentaje de individuos que teniendo predisposición genética desarrolla la enfermedad) es alta y varía tanto para los sexos como para los diferentes órganos. En el varón la penetrancia para el cáncer colorrectal supera el 80%, mientras que en las mujeres es menor (en algunas series, el riesgo para el adenocarcinoma de endometrio supera al colorrectal) 21. La edad promedio de presentación para el CCR es de 45 años y de 50 años para los tumores de endometrio. Otra característica de importancia clínica lo constituye la multiplicidad de los tumores ya sean colónicos (metacrónicos o sincrónicos) o extracolónicos en el momento del diagnóstico.
Si bien el diagnóstico definitivo se basa en la presencia de la mutación en los genes reparadores del ADN, el diagnóstico clínico puede realizarse en base a los antecedentes personales y familiares. Existen diferentes criterios para identificar esta enfermedad pero dado que ninguno de ellos entrega suficiente certeza diagnóstica, deben considerarse fundamentalmente orientativos. Los más utilizados son los denominados «Criterios de Ámsterdam» (Tabla 5). Estos criterios son bastante restrictivos, pero en familias que los cumplen se logra en aproximadamente un 70-80% encontrar la mutación causante. Por otro lado, hay múltiples factores que dificultan su cumplimiento (desconocimiento familiar, fallecimiento temprano por otras causas, familias reducidas, falsa paternidad, entre otros). Por este motivo, es importante conocer las características de la enfermedad para poder sospecharla aun cuando los criterios de Amsterdam no se cumplan. Para fundamentar una sospecha se pueden aplicar los criterios de Bethesda, que son menos estrictos e incluyen un grupo más amplio de personas, pero el rendimiento diagnóstico en el estudio genético es notablemente más bajo (20-30%) (Tabla 6).
CRITERIOS DE AMSTERDAM
| • Criterios de Amsterdam I (historicos): Presencia de CCR • Criterios de Amsterdam II (actuales): Presencia de cualquier cáncer relacionado con el Síndrome • Regla 3-2-1: - Al menos 3 familiares comprometidos - Al menos 2 generaciones consecutivas afectadas - Al menos1 paciente con cáncer bajo 50 años • Poliposis adenomatosa familiar descartada • Confirmación histopatológica de los cánceres |
Fuente: Vasen HFA et al. 7.
CRITERIOS DE BETHESDA
| • Cáncer colorrectal (CCR) diagnosticado en un paciente menor de 50 años de edad. • Presencia de tumores colorrectales sincrónicos, metacrónicos, u otros tumores asociados al síndrome de Lynch, independiente de la edad. • CCR con inestabilidad microsatelital alta (MSI-H) en pacientes menores de 60 años. •Pacientes con CCR y un pariente de primer grado que presentan un tumor asociado al síndrome de Lynch, con uno de los cánceres diagnosticados antes de los 50 años. • Pacientes con CCR con dos o más parientes de primer o segundo grado con un tumor asociado al síndrome de Lynch, independiente de la edad. |
Fuente: Vasen HFA et al. 7.
El término “no polipósico” se adoptó para diferenciarlo del fenotipo clásico de la poliposis adenomatosa familiar (PAF), donde el número de pólipos habitualmente supera el centenar. Sin embargo, al igual que en las otras formas de cáncer colorrectal, la lesión premaligna típica es el pólipo. Al momento del diagnóstico, pueden presentarse pólipos sincrónicos que en general no superan el número de diez. Esto facilita el diagnóstico diferencial con la PAF típica, pero puede dificultar su diferenciación de las formas de PAF atenuadas. Los pólipos se caracterizan por presentarse a una edad más precoz y ser de mayor tamaño que en la población general.
Histológicamente tienden a ser adenomas vellosos con displasia de alto grado. Desde el punto de vista molecular, presentan inestabilidad microsatélite en el 67% de los casos, con frecuente pérdida de expresión inmunohistoquímica de las proteínas MLH1 y MSH2. El potencial maligno de estos pólipos está aumentado. Mientras que en la población general la prevención de un cáncer requiere de 40 a 120 polipectomías y la transformación demanda entre ocho y diez años, en los portadores de esta afección un cáncer se evita cada dos a ocho polipectomías y la transformación maligna se produce en dos a tres años. Llamativamente, los pólipos están distribuidos en todo el colon, sin el predominio proximal que se observa para el adenocarcinoma en el mismo trastorno.
Esto podría explicarse por el desarrollo tumoral a partir de adenomas planos, lesiones que predominan proximalmente y que representan el 50% de los pólipos en esta población. Los adenocarcinomas colorrectales se presentan a una edad promedio de 45 años, aunque algunos portadores los desarrollan a edades más avanzadas (después de los 65 años). Otra característica que lo diferencia de las formas esporádicas es la mayor proporción de lesiones malignas ubicadas en el colon derecho (70%) y una alta incidencia de tumores sincrónicos (18%) y metacrónicos (50% a los 10 años). Estas características demandan una estrategia de vigilancia y tratamiento diferentes. La incidencia de metástasis ganglionares es menor que en las formas esporádicas (35% vs. 65%) 21.
Neoplasias extracolónicasEl espectro de afectación extracolónica es amplio y no muy bien definido. Existe un grupo de órganos con clara asociación (Tabla 7). Tal como acontece con el cáncer colorrectal, se presentan unos diez a quince años antes que las formas esporádicas. El riesgo para los diferentes tumores varía de acuerdo al gen afectado. En relación con las mutaciones del gen MLH1, las mutaciones del gen MSH2 se asocian con un mayor riesgo de tumores extracolónicos (11-42% vs. 48-61%, respectivamente). A su vez, las mutaciones del gen MSH6 se caracterizan por el alto riesgo de cáncer de endometrio (73% vs. 31% del gen MLH1 y 29% del MSH2), presentación más tardía y una menor incidencia de inestabilidad microsatelital. Los portadores de mutaciones en el gen MSH6 tienen un menor riesgo de afectación y suelen afectarse a edades más avanzadas 22.
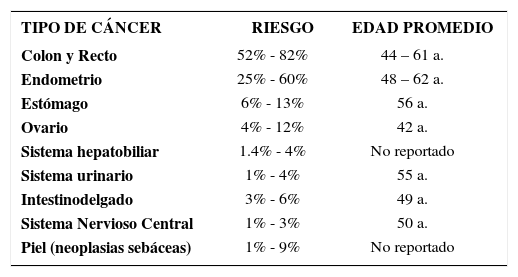
TUMORES ASOCIADOS AL SÍNDROME DE LYNCH
| TIPO DE CÁNCER | RIESGO | EDAD PROMEDIO |
|---|---|---|
| Colon y Recto | 52% - 82% | 44 – 61 a. |
| Endometrio | 25% - 60% | 48 – 62 a. |
| Estómago | 6% - 13% | 56 a. |
| Ovario | 4% - 12% | 42 a. |
| Sistema hepatobiliar | 1.4% - 4% | No reportado |
| Sistema urinario | 1% - 4% | 55 a. |
| Intestinodelgado | 3% - 6% | 49 a. |
| Sistema Nervioso Central | 1% - 3% | 50 a. |
| Piel (neoplasias sebáceas) | 1% - 9% | No reportado |
Fuente: Vasen HFA et al. 7
Las actuales técnicas moleculares permiten identificar las mutaciones que se asocian con el síndrome de Lynch, aunque su sensibilidad no supera el 80%. Debido a su costo relativamente alto, se recomienda efectuar algún tipo de tamizaje previo a la secuenciación. Esto puede efectuarse a través de la determinación de inestabilidad microsatelital o con la de la expresión inmunohistoquímica.
Tamizaje previo al estudio genéticoLa inestabilidad microsatelital idealmente debe determinarse con el panel de Bethesda que implica el uso de 5 marcadores (BAT 25, BAT 26, D2S123, D17S250 y D5S346). Si dos o más marcadores son inestables se clasifica como inestabilidad microsatelital alta.
Otra forma de tamizaje es la determinación de la expresión inmunohistoquímica de las proteínas codificadas por los genes reparadores MSH2, MLH1, MSH6 y PMS2. La inmunohistoquímica es un método mucho más simple y económico que, además, permite determinar cuál es el gen específico alterado. Su desventaja es la menor sensibilidad, ya que no detecta la disfunción de otros genes reparadores. A su vez, es posible que haya expresión inmunohistoquímica casi normal pero que la proteína no tenga capacidad funcional.
La identificación de la mutación patogénica es la única herramienta para confirmar el diagnóstico en el paciente afectado y estimar el riesgo en los familiares consanguíneos. Están indicadas luego de haberse evidenciado inestabilidad microsatelital alta en el tumor o falta de expresión inmunohistoquímica en algunas de las proteínas. También están indicadas en los pacientes con Criterios de Ámsterdam independientemente de los estudios previamente mencionados.
Estudio genéticoActualmente hay disponibles dos técnicas: la secuenciación y la MLPA (Mutliplex Ligation-dependent Probe Amplification). La primera identifica las mutaciones puntuales (missense, non sense, pequeñas deleciones e inserciones) que son el tipo más frecuente, y la segunda identifica mutaciones en grandes deleciones (ej. exones enteros), las cuales son más frecuentes en la afectación del MSH2 y está indicada cuando la secuenciación resulta negativa.
Por su parte, la interpretación de la patogenicidad de las mutaciones es clave y no siempre está claramente definida. Esto es especialmente dificultoso cuando se trata de mutaciones missense (que llevan a una sustitución de un aminoácido) y que se representan hasta en un 30% de las mutaciones identificadas en el gen MLH1. En la base de datos del International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (www.nfdht.nl) se encuentran registradas más de 440 mutaciones patógenas. El 50% de dichas mutaciones afecta al gen MLH1; el 40% al gen MSH2; el 10% al gen MSH6 y el 5% al PMS2.
Otros aspectos importantes de la implementación del diagnóstico genético son aquellos relacionados al impacto psicológico, sociológico y legal. Entre 163 miembros de la Nacional Society of Genetic Counselors, el 91% respondió que aceptaría realizar la prueba genética, pero la mayoría (68%) no lo haría a través de su cobertura médica por temor a la discriminación, y un 57% buscaría ayuda psicológica para sobrellevar los resultados. El riesgo de depresión alcanza al 70% de las personas con resultados positivos. El sentimiento de culpa puede presentarse tanto cuando el resultado es positivo (por la posibilidad de transmitir la predisposición a un descendiente), como cuando resulta negativo (“culpa del sobreviviente”).
La complejidad descripta en la implementación de pruebas genéticas hace necesario que se efectúen solo en el contexto de un asesoramiento genético realizado en el marco de grupos especializados multidisciplinarios.
PREVENCIÓN PRIMARIA Y SECUNDARIARecientemente se ha publicado que la administración de 600mg diarios de aspirina reduce el riesgo de cáncer colorrectal en los portadores sanos 23.
En relación con la prevención secundaria, la vigilancia colorrectal debe comenzarse entre los 20 y los 25 años de edad o 2-5 años antes del familiar más tempranamente afectado, mediante una colonoscopía completa, que deberá repetirse cada uno a dos años. Si bien la única evidencia científica fue obtenida sobre protocolos con endoscopías realizadas cada tres años, la ocurrencia de lesiones en el intervalo de los tres años refuerza la sugerencia de acortar ese plazo a un año. El resto de los órganos con riesgo aumentado carece de una estrategia de eficacia comprobada, aunque existen recomendaciones bien establecidas. En relación al cáncer de endometrio y ovario, se recomienda la realización de ecografía transvaginal y determinación de CA-125 a partir de los 25-35 años, con examen ginecológico cada uno a tres años. Si bien no hay evidencia clara con respecto a las biopsias de endometrio, podría considerarse como una alternativa válida en casos individuales. La vigilancia de otros órganos tampoco tiene comprobada eficacia. Se sugiere, sobre todo en familias con antecedentes de afectación urinaria, la realización de análisis de orina y citológico urinario anual comenzando a partir de los 25-35 años.
Existen distintas posturas con respecto al cáncer del tracto digestivo superior, principalmente para el cáncer gástrico, recomendándose su vigilancia solo en familias con afectación gástrica, mientras que otros sugieren que se realice en forma rutinaria. En todos los casos mediante la realización de una endoscopia digestiva alta con evaluación hasta segunda porción duodenal (eventualmente con extensión hasta yeyuno) a partir de los 30 años cada 2-3 años. En todos los casos con toma de biopsias múltiples de los diferentes sectores del estómago para la detección de inflamación crónica, gastropatía atrófica o metaplasia intestinal y búsqueda de Helicobacter pylori. Aumentando el intervalo ante estudios normales y acortándolo si los hallazgos fueran positivos.
En ciertos casos podrá considerarse la utilización de la capsula endoscópica para el estudio del intestino delgado 24.
TRATAMIENTO QUIRÚRGICOColon
En los portadores sanos, la colectomía profiláctica puede ser presentada como una alternativa a la vigilancia endoscópica anual. Sin embargo, dada la penetrancia incompleta, las alteraciones de la calidad de vida y la morbimortalidad asociadas al procedimiento, y la comprobada eficacia de la colonoscopía para prevenir lesiones avanzadas, no es recomendable. Sin embargo, existen algunas situaciones específicas en las que podría ser considerada: pacientes que no podrán adherirse a la vigilancia mediante estudio completo del colon (colonoscopía) en forma periódica y regular debido a dificultades anatómicas, pobre adherencia a la vigilancia, o trastornos psicológicos que impidan tolerar el miedo a padecer CCR.
Si el paciente desarrolla un adenoma la decisión deberá estar basada fundamentalmente en el tamaño, el número, la frecuencia de la aparición y la posibilidad de resección y vigilancia endoscópica posterior, considerando que en estos pacientes la secuencia adenoma-carcinoma esta acelerada. En presencia de un adenoma no resecable endoscópicamente o un adenocarcinoma, existen dos alternativas: la colectomía total con íleo-recto anastomosis o la colectomía parcial con control endoscópico anual del colon remanente. Si bien la evidencia es limitada, la recomendación de expertos es la resección ampliada, ya que la acelerada progresión pólipo-cáncer hace que el riesgo de padecer lesiones en el segmento remanente esté presente aun con controles endoscópicos anuales. Sin embargo, las resecciones más limitadas tendrían indicación en los pacientes añosos, en quienes el riesgo de un tumor metacrónico es menor (por su expectativa de vida), y los trastornos funcionales pueden ser mayores. Por estas razones, la elección entre uno y otro tipo de colectomía, debe ser hecha considerando circunstancias individuales (comorbilidades, hábitos evacuatorios, aceptación y facilidad para completar estudios endoscópicos, entre otros).
Por otra parte, debe recordarse que, aun cuando se realiza una colectomía total, el riesgo de presentar un cáncer de recto alcanza el 12% a los diez años, por lo que debe efectuarse una vigilancia endoscópica anual. Cuando la lesión se presenta en el recto, las opciones son la proctocolectomía total con reservorio ileoanal o la resección anterior y control endoscópico. Teniendo en cuenta la mayor morbilidad postoperatoria inmediata y alejada de la primera opción, nuestra recomendación es la resección conservadora.
Ovario y útero
El alto riesgo de cáncer de endometrio y ovario, así como la deficiencia de las estrategias de prevención justifican la histerectomía con ooforectromía bilateral profiláctica. Esta alternativa es claramente aceptable en mujeres post menopáusicas o que no desean procrear y deben ser operadas por un CCR. En las mujeres que no están afectadas (ya sea por haber sido sometidas a cirugía colorrectal o no desarrollaron un CCR), esta alternativa es también válida aunque implica una intervención quirúrgica exclusivamente para este fin.
CONCLUSIÓNLos síndromes hereditarios descritos causan cáncer colorrectal y extracolónicos en los individuos afectados. Es importante, reconocer la eventual presencia de un síndrome hereditario en un paciente con cáncer, evaluar detalladamente la historia familiar del paciente, y de esta manera fundamentar un eventual estudio genético. Una vez identificada la mutación, se pueden identificar los portadores de la mutación en el grupo familiar, y de esta manera establecer pautas de vigilancia para cada individuo en riesgo, con el fin de prevenir el desarrollo de un cáncer asociado a la mutación, o al menos detectarlo de manera precoz, para poder ofrecer un tratamiento eficiente.
Los autores declaran no tener conflictos de interés en relación a este artículo.