







Los tumores primarios malignos del sistema nervioso central (SNC) son los tumores sólidos más comunes de la infancia y después de las leucemias, ocupan el segundo lugar en frecuencia. El tratamiento de los tumores cerebrales es complejo por la diversidad histológica de las lesiones y la tendencia de la mayoría de ellos a diseminar en el neuroeje precozmente en el curso de la enfermedad. Además, pueden producirse secuelas importantes secundarias a las intervenciones terapéuticas.
Aunque se han obtenido mejorías en las tasas de sobrevida, estas no son comparables con los avances logrados en otros tumores infantiles en las últimas tres décadas. Esto puede explicarse en parte, a la falta de conocimiento sobre el comportamiento biológico de estos tumores, lo cual los hace todavía un desafío para el equipo médico tratante.
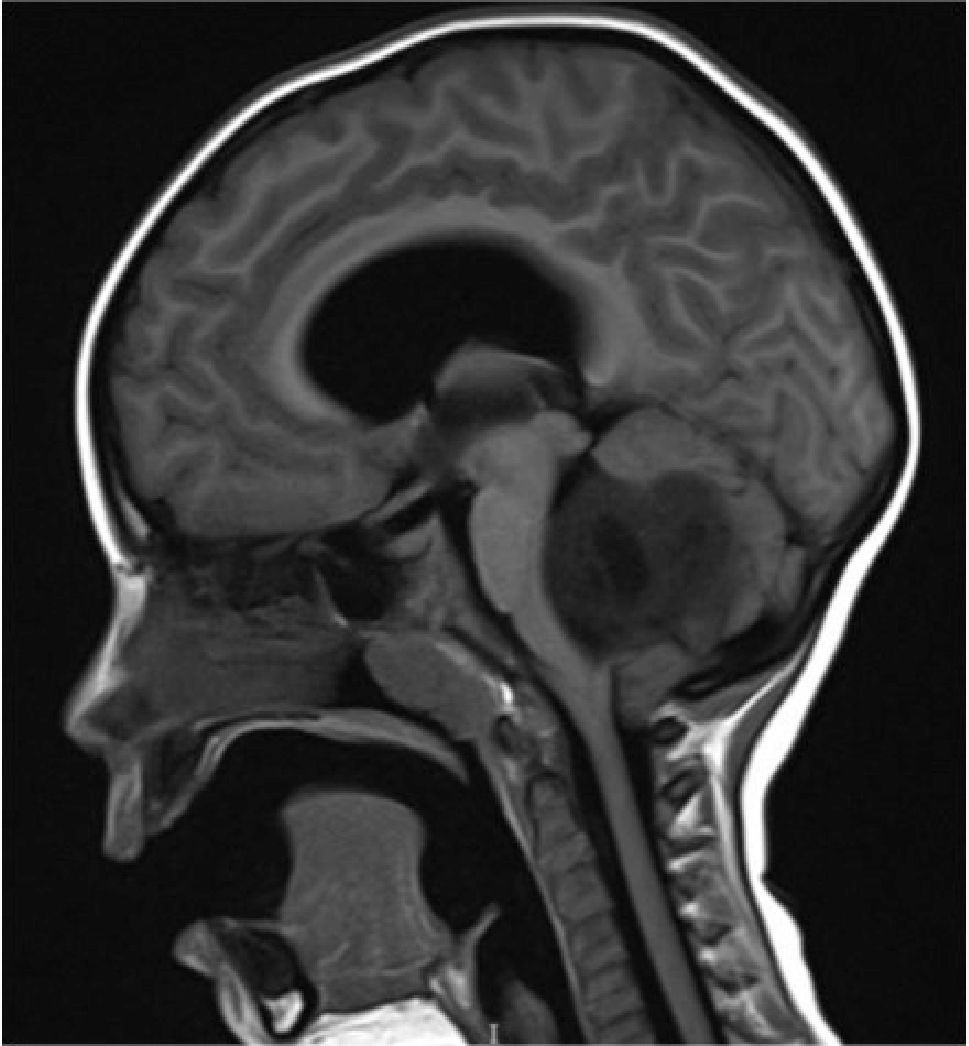
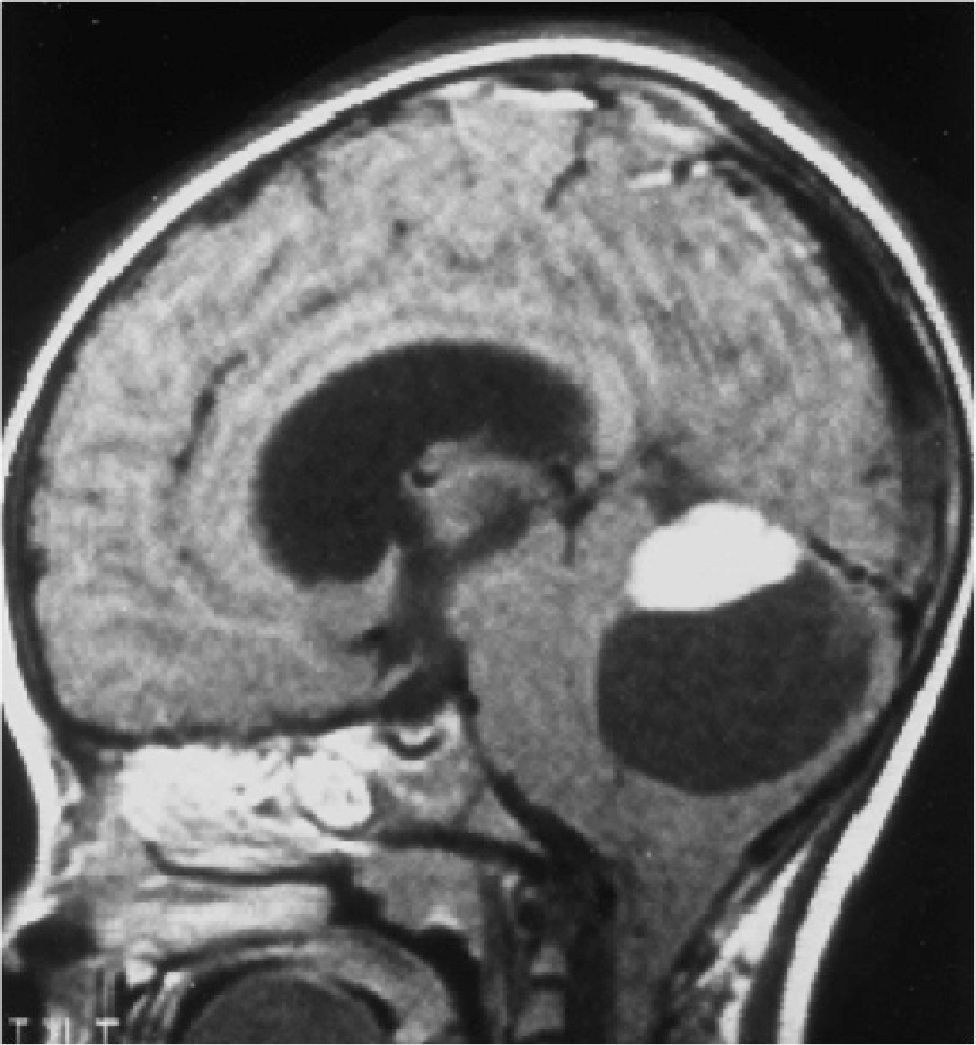
Los tumores de la fosa posterior son aquellas neoplasias primarias o secundarias ubicadas en la región infratentorial, o sea, bajo la tienda del cerebelo. Estos tumores o neoplasias pueden originarse en cualquier estructura de esta región. La fosa posterior es pequeña, en ella se encuentra el 25% del contenido intracraneal, pero contiene estructuras vitales como el tronco cerebral. Así, los tumores de fosa posterior son letales por compresión e hipertensión secundaria, obstrucción de la circulación de líquido cefalorraquideo y por destrucción del parénquima por infiltración (Figura 1).
En los niños predominan los tumores infratentoriales primarios, los que corresponden al 50-70% del total de tumores intracraneanos. En los adultos, los tumores predominan en el compartimiento supratentorial y en la fosa posterior son más frecuentes las metástasis.
En esta revisión se analizarán aspectos epidemiológicos, patogenia y biología molecular clínicos y neuroradiológicos en general de los tumores de fosa posterior y en particular se revisarán los avances en biología molecular y tratamiento de los tumores más frecuentes de la zona; méduloblastoma, ependimoma, astrocitoma de bajo grado y los tumores de tronco cerebral.
Malignant primary tumors of the central nervous system (CNS) are the most common solid tumors of childhood and, after leukemias, occupy second place in frequency. The treatment of brain tumors is complex because of the histological diversity of the lesions and the tendency of most of them to spread in the neuraxis early in the course of the disease. In addition, significant sequelae secondary to therapeutic interventions may occur.
Although improvements in survival rates have been obtained, they are not comparable with advances in other childhood tumors over the past three decades. This may be explained in part by the lack of knowledge about the biological behavior of these tumors, which makes them still a challenge for the treating medical team.
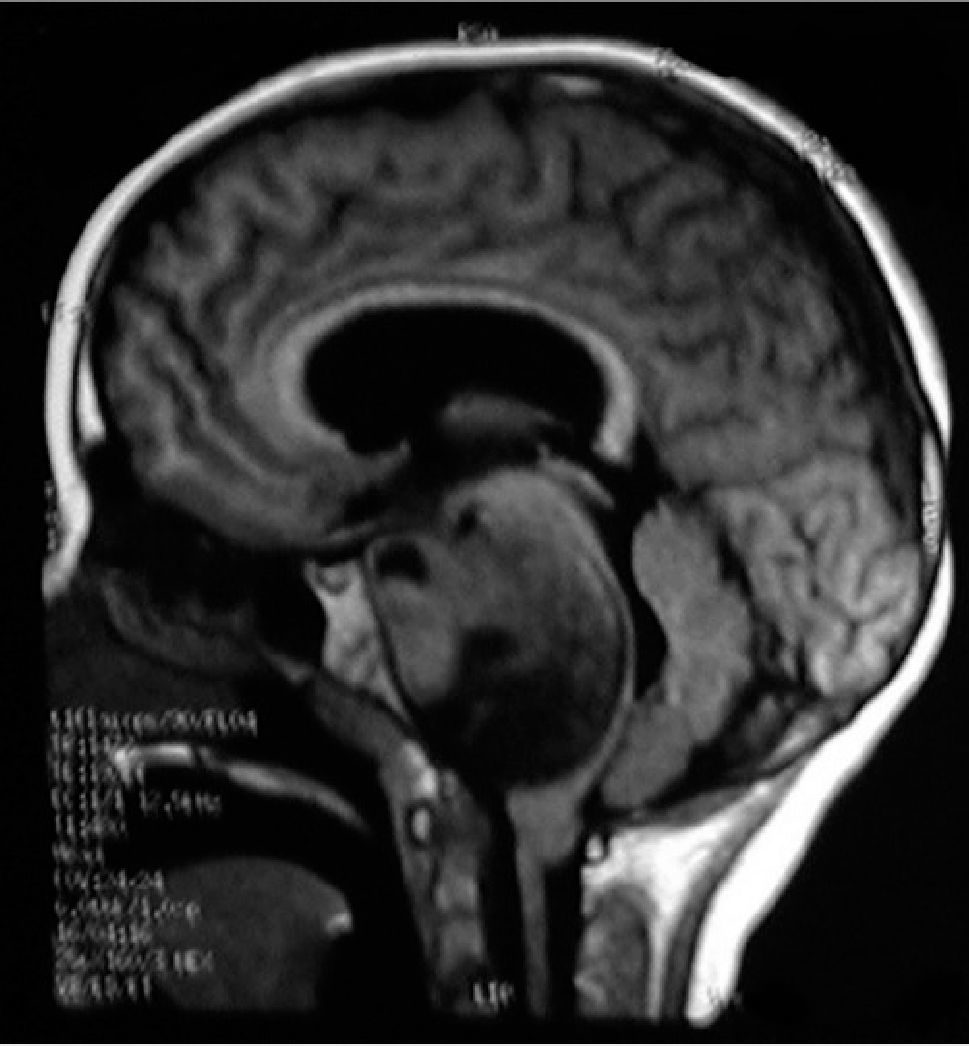
Tumors of the posterior fossa are those primary or secondary neoplasias located in the infratentorial region, that is, under the cerebellum tent. These tumors or neoplasms can originate in any structure of this region. The posterior fossa is small, on it is 25% of the intracranial content, but it contains vital structures like the brainstem. Thus, posterior fossa tumors are lethal by compression and secondary hypertension, obstruction of cerebrospinal fluid circulation, and destruction of the parenchyma by infiltration (Figure 1).
In children, primary infratentorial tumors predominate, corresponding to 50-70% of all intracranial tumors. In adults, tumors predominate in the supratentorial compartment and in the posterior fossa, metastases are more frequent.
This review will describe epidemiological, pathogenesis, clinical, molecular biology and neuroradiological aspects in general of tumors of the posterior fossa and in particular we will review the advances in molecular biology and treatment of the most frequent tumors in the area; medulloblastoma, ependymoma, low grade astrocytoma, and brain stem tumors.
Desde 1997, el Programa Infantil Nacional de Drogas Antineoplásicas (PINDA), dependiente del Ministerio de Salud, incluye en sus Protocolos el tratamiento de estos tumores, comenzando entonces con el registro de pacientes afectados por esta patología.

En el registro Nacional de Cáncer Infantil de Chile, (RENCI-Quinquenio 2007-2011) publicado en 2016, de aproximadamente 480 casos nuevos de cáncer en menores de 15 años al año en nuestro país, el 15.8% de ellos son tumores cerebrales 1. Esta cifra es similar a la de otros países 2. En Estados Unidos corresponden al 16.6% de todos los tumores en pacientes menores de 20 años.
Se ha descrito en la literatura un aumento de la incidencia de estos tumores en las últimas décadas, lo que corresponde en realidad a una mejoría en el diagnóstico gracias al desarrollo y mayor disponibilidad de métodos de imágenes y a una sospecha clínica más precoz. Todo esto hace plantear que existía subdiagnóstico en este tipo de tumores.
La incidencia de los tumores cerebrales presenta un alza en la primera década de la vida y luego un segundo peak en la edad adulta, alrededor de los 70 años, que corresponde principalmente a metástasis tumorales. En niños los cánceres más comunes que hacen metástasis al cerebro son: leucemias, linfomas, sarcomas, neuroblastomas y tumores de células germinales.
En la etapa pediátrica la localización del tumor se relaciona con la edad; en los primeros 3 años de vida los tumores supratentoriales son más comunes, los infratentoriales predominan en los pacientes de 4 a 10 años de edad. En los niños mayores de 10 años se desarrollan tumores en ambas localizaciones con igual frecuencia.
Las diferencias en las tasas de sobrevida de niños y adultos reflejan ampliamente las diferencias histológicas que predominan en estos dos grupos, por ejemplo la tasa de sobrevida en menores de 15 años para tumores del sistema nervioso central es de un 58.7%, mientras que para el tumor más común en adultos como es el glioblastoma multiforme es de 6%.
PATOGENIA Y BIOLOGÍA MOLECULARLos avances recientes en la biología molecular han permitido desarrollar el estudio de los tumores cerebrales de manera mucho más especifica que la histología tradicional. El análisis molecular de ciertos marcadores genéticos provee información adicional en cuanto a la clasificación y pronóstico de los tumores en general. En el caso de los tumores cerebrales hay algunos estudios que sugieren que el patrón de alteraciones genéticas puede ser distintivo en los pacientes pediátricos comparado con los adultos. En pediatría en especial se ha avanzado en el estudio genético y molecular de la tumorogénesis del méduloblastoma 3 en forma significativa. El estudio genético por microarray, técnica que permite el análisis de DNA en forma simultánea de varios tipos de tumores, demuestra que estos tumores tienen un comportamiento diferente al de los otros PNET, por lo tanto, este tipo de estudio, asociado a los criterios clínicos e histológicos, permite la estratificación de los pacientes en distintos grupos de riesgo para diferentes protocolos de tratamiento 4.
Una serie de síndromes genéticos se asocian con ciertos tumores cerebrales, incluyendo la neurofibromatosis tipo I (gliomas de vía óptica), esclerosis tuberosa (astrocitomas, ependimomas). En el caso de los méduloblastomas (MB) la mayoría ocurren en forma esporádica, sin embargo, existen tres síndromes bien conocidos que se le asocian, el síndrome de Gorlin en el cual se ha descrito mutaciones en los siguientes genes, PTCH15,6, PTCH27, SUFU(8) que llevan a una desregulación de la vía Sonic Hedgehog(SHH). El síndrome de Turcot que se caracteriza por una desregulación de la vía de las beta cateninas (WNT/b cateninas) y el síndrome de Li Fraumeni caracterizado por una mutación del p53 lo cual predispone a cáncer.
En ependimomas el genoma es balanceado en más del 50% de los casos y la caracterización genética ha permitido definir distintas subclases según ARNm característico. No existe una relación síndrome familiar/ependimoma y las anomalías citogenéticas son múltiples.
En gliomas de bajo grado existen múltiples subtipos con mutación del oncogen BRAF que activa la vía del MAPK, esto se ha estudiados en síndromes genéticos asociados a gliomas de bajo grado como la esclerosis tuberosa y la Neurofibromatosis tipo 1.
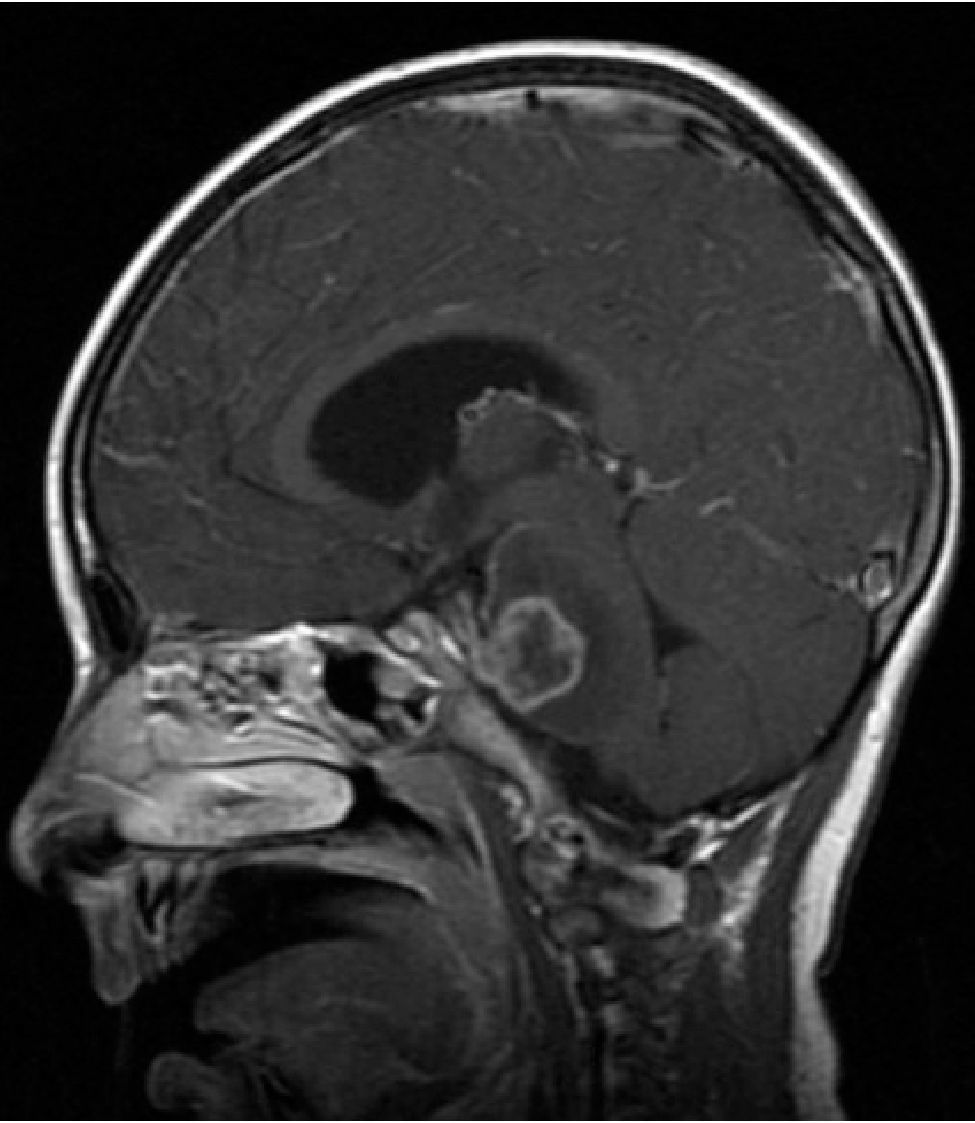
NEUROIMÁGENESLos avances en neurorradiología han facilitado considerablemente el diagnóstico de los tumores encefálicos. El rol de las imágenes no radica en la predicción del tipo de tumor, que habitualmente es inexacta, a excepción de tres tipos de tumores: los tumores de células germinales secretores, los gliomas infiltrantes difusos de tronco (Figura 2) y los gliomas de vía óptica en los pacientes con Neurofibromatosis tipo I. La utilidad de la imagenología se centra en: la detección del tumor, su localización y la demostración de efectos secundarios, tales como herniación, hidrocefalia y hemorragia. Las imágenes deben ayudar en la caracterización de la textura y márgenes de la lesión y en la detección de extensiones o infiltraciones tumorales; todos estos factores influencian la posibilidad de resección del tumor y proveen al cirujano de un mapa resectivo 9,10.

Actualmente la Tomografía Axial Computada (TAC) y la Resonancia Magnética (RM) son de amplia disponibilidad. Estudios han demostrado que la RM es más sensible y específica que la TAC en la detección de procesos expansivos intracraneales (92% y 99% respectivamente para RM versus 81% y 92% respectivamente para TAC). Estudios de costo-efectividad que analizan las estrategias de imágenes para niños con cefalea sugerente de ser secundaria a tumor cerebral (cefaleas de menos de 6 meses de duración, con algún otro predictor clínico: déficit neurológico focal o edema de papila) demuestran que el estudio con RM es más efectivo.
La TAC es superior para la detección de calcificaciones, lesiones de base de cráneo y calota asociadas y hemorragia aguda. Por otro lado, la RM permite un mejor estudio de los tejidos blandos, de las lesiones isodensas al TAC, de la captación de contraste y de hallazgos asociados tales como edema, hemorragia (excepto la aguda) e infarto. Además, la RM gracias a su visión en múltiples planos, permite una mejor distinción entre lesiones intra y extra axiales, mejor localización en el espacio y mejor identificación de las estructuras comprometidas por el tumor. La RM evalúa mejor las lesiones de fosa posterior y de la porción inferior de los lóbulos frontal y temporal.
La espectroscopía por RM del protón, es un estudio suplementario que permite distinguir tumor de otras lesiones. Algunos estudios de espectroscopía por RM, caracterizando propiedades tisulares han podido predecir el tipo de tumor con un 95% de exactitud. Estudios de medicina nuclear tales como el SPECT (Single photon emission computed tomography) y PET (Positron emisssion tomography), son útiles especialmente en la diferenciación de recurrencia tumoral y necrosis post irradiación.
Ocasionalmente el estudio angiográfico y la embolización por vía arterial del tumor son útiles antes de la resección quirúrgica, para minimizar la hemorragia perioperatoria.
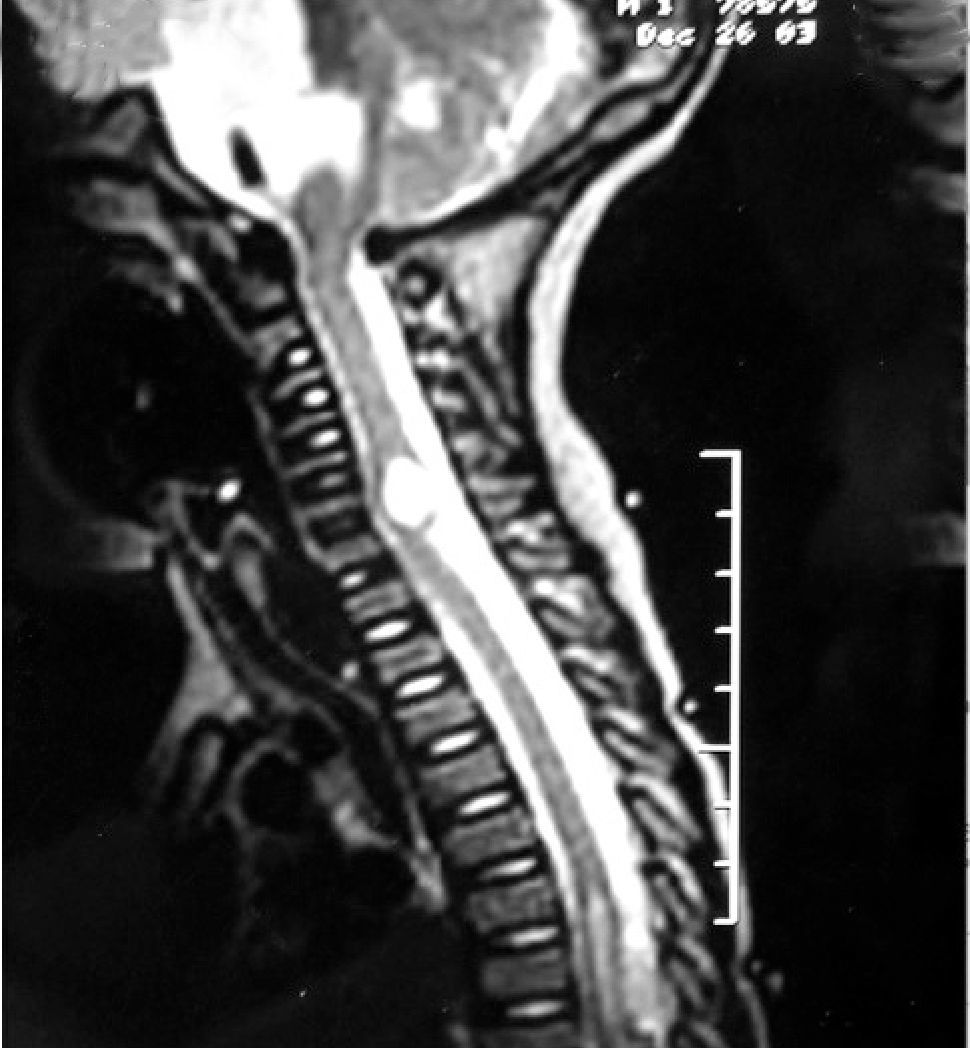
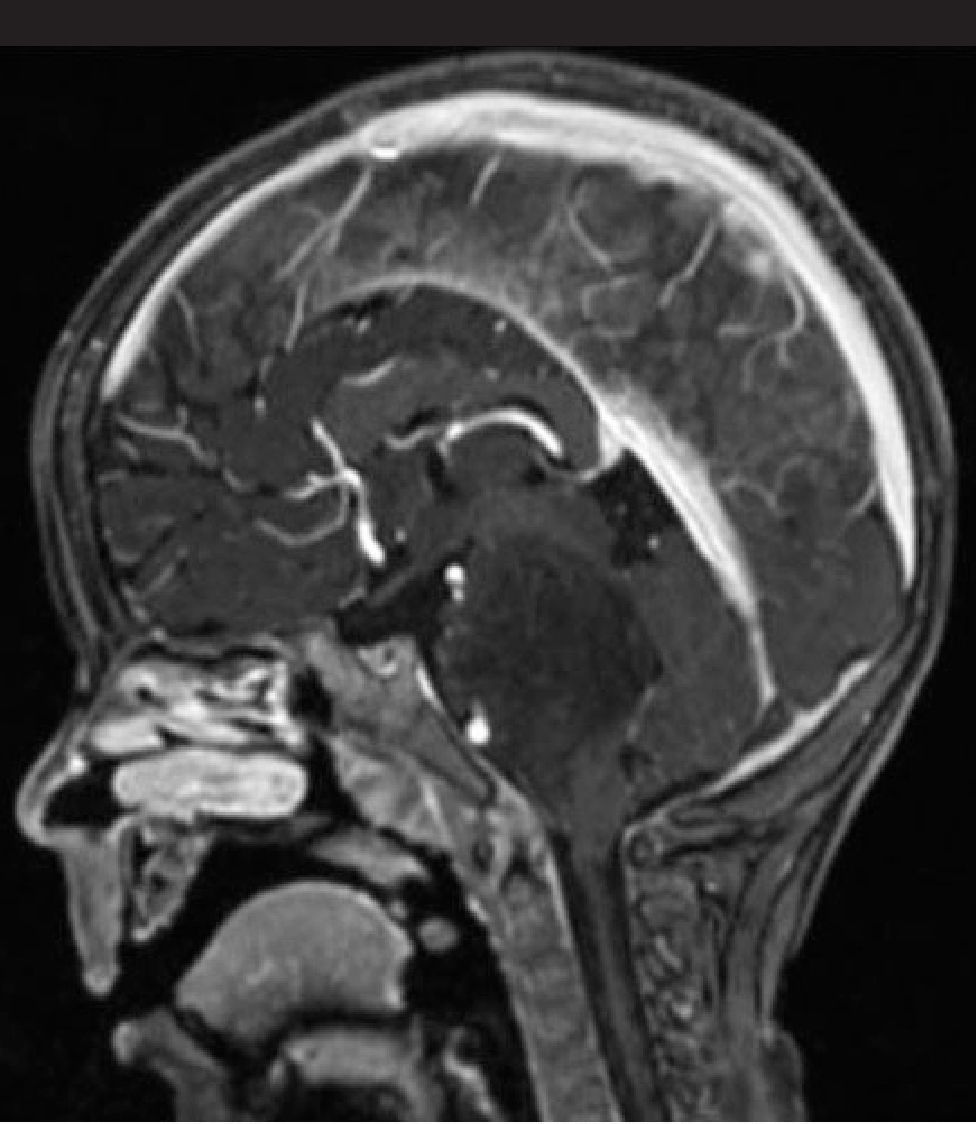
Una RM cráneoespinal con gadolinio es indispensable para la evaluación de la siembra tumoral (Figura 3). Esto es especialmente importante en Meduloblastoma, otros tumores neuroectodérmicos primitivos, tumores de células germinales y gliomas de alto grado o indiferenciados. Este estudio idealmente debe realizarse al momento del diagnóstico, en forma preoperatoria, dado que durante el período postoperatorio, la presencia de coágulos sanguíneos y restos tisulares pueden simular metástasis.

El rol de la imagenología en la fase postoperatoria y en el seguimiento, radica en detectar las complicaciones de la terapia y la presencia de tumor residual o recurrencia. Una imagen postquirúrgica debería obtenerse inmediatamente después de la cirugía. Retardar este estudio a más de 72 horas de ella, puede inducir a error debido a que la captación reactiva del contraste en el sitio operatorio puede simular tumor residual.
No existe consenso sobre la frecuencia y oportunidad del estudio de imágenes para la detección de recidivas en pacientes asintomáticos.
CUADRO CLÍNICOLas principales estructuras anatómicas de la fosa posterior son el tronco cerebral, el cerebelo y los pares craneananos. En consecuencia los síntomas asociados a tumores localizados en esta región se relacionan a estas estructuras. De manera de sistematizar podemos agrupar los signos y síntomas en cuatro categorías principales: 3–10
- 1.
Síndrome de hipertensión intracraneal
- 2.
Signos neurológicos focales
- 3.
Convulsiones
- 4.
Meningismo
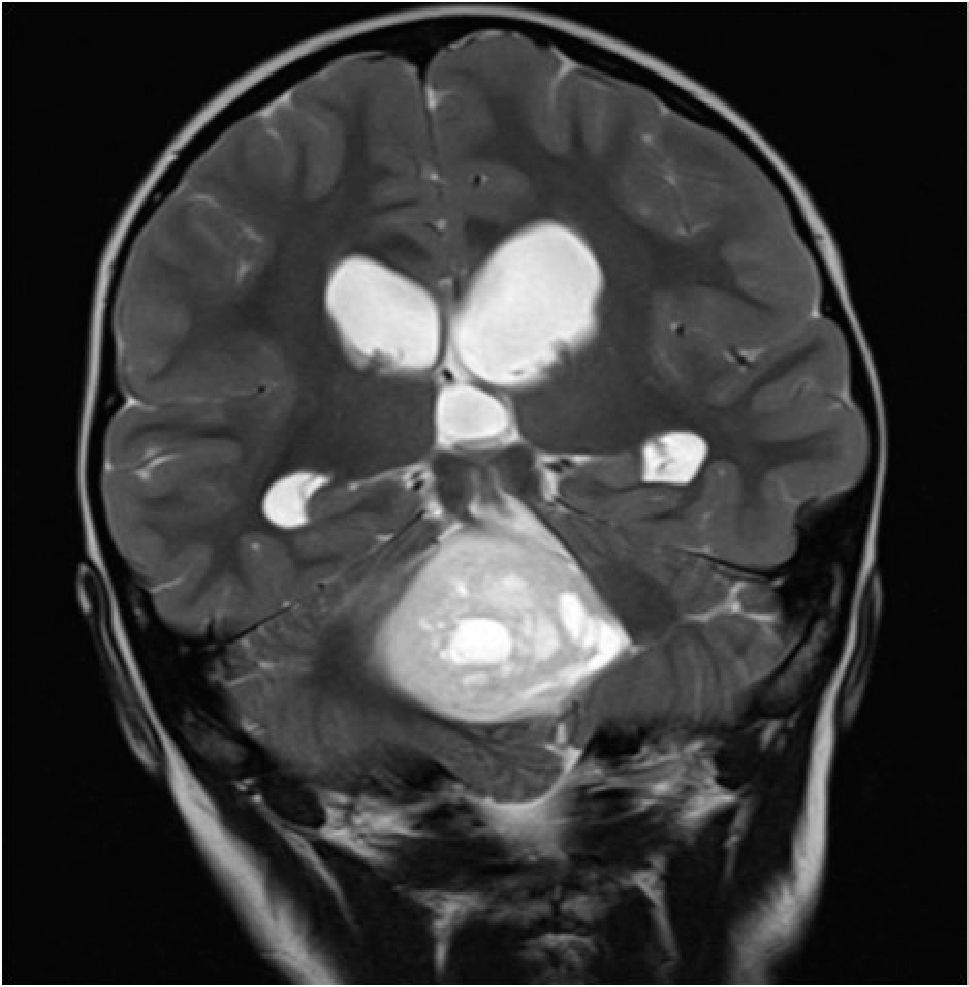
Constituye el síndrome más común de presentación en los niños con tumores de la fosa posterior, siendo el sello de esta enfermedad. Cualquier proceso expansivo que obstruya la circulación de LCR producirá hipertensión intracraneal por aumento del compartimiento de LCR, incluso sin hallazgos de lateralización (Figura 4).

Existirá cefalea, lejos el signo más frecuente en los tumores de fosa posterior, que es de comienzo insidioso para luego hacerse más frecuente e intensa. Puede presentarse en la mañana al levantarse, o en la madrugada siendo razón suficiente para despertarlo temprano. En el transcurso del día la cefalea va disminuyendo y se alivia marcadamente después del vómito. Se alivia muy poco con los analgésicos comunes. Puede extenderse a toda la cabeza (holocraneana) o, en las etapas iniciales, localizarse en la región suboccipital-occipital. Se exagera en circunstancias que hacen variar la distribución del LCR, por ejemplo, tos, estornudos, defecación, cambios bruscos de posición de la cabeza y del cuerpo. Generalmente, la cefalea dura semanas o meses previos al diagnóstico.
Asociado a la cefalea existen frecuentemente los vómitos, los cuales pueden ser intensos e intratables, especialmente cuando existe infiltración del piso del cuarto ventrículo comprometiendo el núcleo del vago. Eventualmente puede comprometer el estado nutricional del menor con anorexia y disminución de peso, haciendo más difícil el diagnóstico 11.
También encontraremos signos oculares. Se puede presentar diplopia con estrabismo por compromiso no localizatorio de uno o ambos VI pares craneales, por compresión hipertensiva del nervio en el borde libre de la tienda del cerebelo. Clínicamente se traduce en paresia del correspondiente músculo recto externo. El paciente describirá su diplopia como doble imagen en el plano horizontal y tendrá paresia de la mirada lateral hacia el lado afectado, quizás con rotación compensatoria de la cabeza hacia el globo ocular afectado 12.
Con frecuencia se encontrará edema papilar. Su diagnóstico se realiza examinando el fondo de ojo. El edema de papila generalmente lo encontramos presente al momento del diagnóstico. Debemos tener presente que el edema papilar mantenido puede causar ceguera por atrofia de la papila. Es importante, además, diferenciarlo del pseudoedema. Generalmente el edema de papila es más acentuado en los tumores de fosa posterior de la línea media, que en aquellos lateralizados.
En los niños, en que existen suturas permeables, se evidenciará un aumento del perímetro craneal, al igual que el aumento de tensión de la fontanela anterior o bregmática, como otros signos de hipertensión intracraneal.
Signos neurológicos focalesEn el compromiso cerebeloso distinguiremos manifestaciones clínicas características para el compromiso de línea media y para el compromiso lateral.
El compromiso vermiano o de línea media se caracteriza por trastornos de la estación de pie (postura y equilibrio) y de la motilidad de los miembros inferiores, con asinergia de tronco y tendencia a la retropulsión. Por lo tanto, existe ataxia de tronco y de la marcha. Esta última es de base aumentada y tambaleante. La marcha en tandem es imperfecta. El paciente estando de pie se balancea en todas direcciones. En posición supina no hay alteración en las extremidades. Puede coexistir nistagmus por compromiso de las vías vestíbulo-cerebelosas. Por lo tanto, es un síndrome de predominio estático y de localización generalmente bilateral.
El compromiso lateral o hemisférico se caracteriza por la incoordinación motriz de las extremidades, manifestado como dismetría y disdiadococinesis. Se agrega hipotonía muscular, hiporreflexia y lateropulsión, menos pronunciado en niños que en adultos. Por lo tanto, es un síndrome de predominio cinético y homolateral a la lesión.
Si las conexiones cerebelosas son las comprometidas, se observarán síndromes cerebelosos asociados a compromiso de otras vías, como manifestaciones sensitivas, motoras o de pares craneanos.
Como se mencionó anteriormente también se puede encontrar nistagmus y vértigo. El nistagmus se encuentra por lo general al momento del diagnóstico. Generalmente es en la mirada horizontal, ocasionalmente en la mirada vertical. Esto se debe al compromiso de los pedúnculos cerebelosos inferiores, hidrocefalia y/o compromiso del tronco cerebral. El vértigo se encuentra generalmente asociado al nistagmus y puede ser difícil de diagnosticar en niños. Por lo general, el niño permanece de un lado u otro, en posición fetal y con los ojos cerrados.
El compromiso de los pares craneales se puede manifestar en los tumores de tronco, en los tumores vermianos o intraventriculares que infiltran el piso del cuarto ventrículo, y en aquéllos tumores que comprometen el ángulo pontocerebeloso.
En los tumores de tronco cerebral podemos encontrar compromiso de varios núcleos de pares craneales en forma uni o bilateral. Cuando el tumor infiltra el piso del cuarto ventrículo, hay compromiso de aquellos que están inmediatamente debajo, como los núcleos del VI y VII par, núcleos vestibulares, hipogloso y vago. Cuando el tumor compromete el ángulo pontocerebeloso, se encontrará compromiso del nervio auditivo, trigémino y facial periférico del mismo lado del tumor 13–15.
ConvulsionesSe presenta rara vez en los tumores de fosa posterior o infratentoriales. Generalmente se describe como “ataque cerebeloso” y no representa una crisis epiléptica propiamente tal, sino más bien una situación de alarma y emergencia, de pronóstico grave. Durante estos episodios el paciente adopta una posición de descerebración, incluso opistótono pronunciado, con inconciencia, bradipnea, bradicardia, hipertensión arterial. Generalmente se debe a hidrocefalia aguda con isquemia de tronco, por lo que la descarga neuronal se origina en la sustancia reticular 16.
Si realmente existe una crisis epiléptica, uno debería pensar en una siembra subaracnoidea sobre la convexidad de los hemisferios cerebrales.
MeningismoGeneralmente la rigidez de nuca denota irritación meníngea por sangre en el espacio subaracnoideo o detritus purulento, como ocurre en los pacientes con hemorragia subaracnoidea o meningitis bacteriana, respectivamente. Es importante tener en cuenta que la rigidez de nuca también puede deberse a un proceso expansivo de la fosa posterior. El paciente presentará un cuello rígido cuando camina, o inclinación de la cabeza hacia un lado, o resistencia a la flexión pasiva del cuello. La rigidez de nuca y dolor representa una irritación y estiramiento de la duramadre, especialmente a nivel de la unión cérvico-medular, por herniación de las amígdalas cerebelosas o tejido tumoral.
Ocasionalmente el meningismo puede manifestarse como una escoliosis dolorosa o radiculopatía, indicando una siembra tumoral hacia el canal espinal, que es más frecuente en el meduloblastoma y en el ependimoma 17.
TUMORES MÁS FRECUENTES EN LA FOSA POSTERIOR EN EDAD PEDIÁTRICAMeduloblastomaLos tumores cerebrales son los tumores sólidos más frecuentes en la edad pediátrica y el meduloblastoma (MB) es el tumor maligno más común. Su peak de incidencia es entre los 5 y 10 años de edad, aunque puede ocurrir en forma congénita y también afectar a pacientes adultos. Es un tumor que predomina en el sexo masculino en una relación de 2:1.
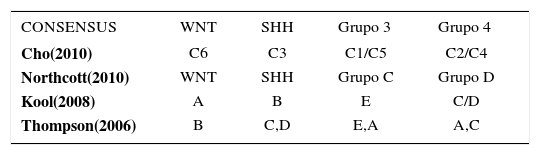
El tumor se origina en un 90% en el vermis (Figura 5) y en un 10% en el hemisferio cerebeloso. En los adolescentes y adultos el tumor tiende a ubicarse lateralmente en el hemisferio cerebeloso, pudiendo inducir una respuesta proliferativa de las leptomeninges (variedad desmoplástica). La clasificación de la OMS clásica dividía el MB es cinco subgrupos; clásico, anaplásico, de células grandes, desmoplástico y de extensa nodularidad. Un consenso de los avances en estudios moleculares permiten que actualmente el MB se pueda clasificar en cuatro subtipos moleculares, WNT, SHH, Grupo 3 y Grupo a (Tabla 1) 4.

Clínicamente se presenta con un síndrome de hipertensión intracraneal asociado a síndrome cerebeloso de línea media o hemisférico. Puede existir compromiso de pares craneales bajos por infiltración del piso del cuarto ventrículo. Ocasionalmente puede presentarse como hemorragia tumoral, por ser un tumor vascularizado. Es el tumor que con más frecuencia puede dar metástasis espinales o supratentoriales por siembra subaracnoidea. Alrededor de un tercio tiene metástasis de las leptomeninges y menos del 10% metástasis extracraneales.
El tratamiento es quirúrgico. Se complementa con quimioterapia y radioterapia craneoespinal. Se realiza estudio postoperatorio con RM cerebral y espinal para evaluar grado de resección tumoral y presencia de metástasis, la biopsia definitiva con su estudio molecular permitirá diseñar un tratamiento complementario óptimo con radioterapia y/o quimioterapia.
El Meduloblastoma es un tumor que tiende a recurrir con frecuencia, con un 70% dentro de los dos primeros años y con mayor frecuencia en la misma fosa posterior. La sobrevida es mala en general, siendo un 50 a 70% a los cinco años y de un 40 a 50% a los diez años. Clásicamente se consideraban factores de mal pronóstico: la edad menor de tres años, presencia de metástasis y un volumen tumoral remanente importante posterior a la cirugía, pero actualmente con la caracterización por subgrupos moleculares se puede anticipar con mucha mayor precisión el comportamiento biológico del tumor y por lo tanto, definir una mejor terapia y pronóstico.
EpendimomaEl ependimoma representa el 10 a 15% de los tumores de la fosa posterior, es el tercer tumor más frecuente en la infancia, su grado de malignidad 18 se asocia a la edad del niño. En menores de 3 años se observa principalmente la variedad anaplásica, por el contrario, en niños mayores de 10 años hay una mayor frecuencia de la variedad bien diferenciada.
Es un tumor que se origina en las paredes del epéndimo del 4° ventrículo, es polilobulado, invade progresivamente el espacio subaracnoídeo extendiéndose hacia el ángulo pontocerebeloso y el canal raquídeo cervical (Figura 6).

El modo de presentación clínica es dependiente de la edad. En el lactante se presentará como una hidrocefalia de evolución rápida, en el niño mayor, el cuadro clínico se instalará más lentamente, con vómitos aislados, signos cerebelosos leves a moderados y una hipertensión endocraneana tardía. Es de una gran heterogeneidad en su edad de presentación, localización, grado histológico y comportamiento clínico.
Su genoma también es heterogéneo y diferentes estudios han mostrado cohortes de tumores con frecuentes alteraciones cromosómicas y otros con un genoma balanceado. La caracterización genética ha permitido definir distintas subclases según ARNm característico. Witt et al. han descrito dos subgrupos diferentes en dos series independientes de tumores de fosa posterior. Denominados subgrupos A y B, el subgrupo A incluye solo ependimomas de fosa posterior y el subgrupo B incluye ependimomas de fosa posterior y espinales. Los pacientes del subgrupo A son menores (mediana 2.5 años) con tumores localizados en forma lateral y con un genoma balanceado. Tienen mayor incidencia de recurrencia, metástasis y se asocian a un peor pronóstico.
Los pacientes del subgrupo B son mayores (mediana 20 años), con tumores de la línea media y alto porcentaje de variabilidad genética, incluyen diferentes anomalías citogenéticas como pérdida de cromosoma 1, 2, 3, 6, 8, 10, 14q, 17q, 22q, y ganancia en cromosomas 4, 5q, 7, 9, 11, 12, 15q, 18, 20, and 21q, estos pacientes tienen un mejor pronóstico de sobreviva 19,20 (Tabla 2).
SUBGRUPOS EPENDIMOMA
| Grupo A | Grupo B | |
|---|---|---|
| Perfil genético | Perfil genómico balanceado | Pérdida de cromosoma 1, 2, 3, 6, 8, 10, 14q, 17q, 22q Ganancia en cromosomas 4, 5q, 7, 9, 11, 12, 15q, 18, 20 y 21q |
| Hallazgos clínicos | Edad media presentación 2.5 años Localización lateral 67% Antígeno marcador LAMA2 | Edad media presentación 20 años Localización línea media 95% Antígeno marcador NELL 2 |
| Pronóstico | Recurrencia a 5 años 56% metástasis frecuentes sobrevida a 5 años 69% | Recurrencia a 5 años 25% sobrevida a 5 años 95% |
Tabla modificada » Medicine Oncology » “Evolution of the Molecular Biology of Brain Tumors and the Therapeutic Implications”, book edited by Terry Lichtor, ISBN 978-953-51-0989-1, Published: February 27, 2013 under CC BY 3.0 license. © The Author(s). Chapter 20.
Las neuroimágenes son fundamentales en la identificación y caracterización de los ependimomas de fosa posterior, tanto en el diagnóstico, planificación prequirúrgica, y evaluación del tratamiento con el grado de resección post operatorio y eficacia de la radioterapia y quimioterapia. La imagen de resonancia magnética (RM) y la tomografía axial computada (TAC) son los métodos más utilizados. Estas técnicas detectan las lesiones con su diferente densidad y señal de intensidad con respecto al tejido cerebral normal, además del efecto de masa con diferentes grados de distorsión de las estructuras normales. La RM es superior al TAC en la caracterización y el seguimiento del tumor post terapia, dado que permite una mejor capacidad de contraste de los diferentes tejidos blandos y la capacidad de adquirir imágenes de manera multiplanar, además de prevenir los efectos deletéreos de la radiación ionizante del TAC. Otras modalidades avanzadas de RM como la difusión, perfusión y la espectroscopía entregan información del comportamiento funcional del tumor, como su celularidad, hemodinamia y metabolismo lo cual agrega información valiosísima en la caracterización del tumor. Toda la información imagenológica será fundamental para la planificación de una cirugía cuyo objetivo es la resección lo más completa posible.
El tratamiento será esencialmente quirúrgico y siempre debe tener como objetivo la extirpación total ya que esto se asocia en forma directa a un mejor pronóstico. El factor más determinante de la sobreviva de la enfermedad es la extensión de la resección. Esto genera una gran responsabilidad en el neurocirujano para lograr una máxima y segura extirpación. La sobrevida a 5 años es de 67-80% en pacientes con una extirpación total o gross total resection (GTR) y la sobrevida libre de progresión de la enfermedad es de 51-75% 21–24. Después de una resección subtotal la sobrevida a 5 años es de un 22-47% y la sobrevida libre de progresión es de un 0-26%.
Según el grado histológico y la edad del paciente, en la mayoría de los casos se complementará la cirugía con un tratamiento coadyudante. La cirugía sola es suficiente en ependimomas de bajo grado localizados en la médula y supratentoriales. En el resto de los casos el ependimoma deberá recibir terapia complementaria. Actualmente los avances de la radioterapia han permitido ampliar el espectro de pacientes hasta niños de 1 año con tumores de fosa posterior 25,26 donde se han descrito efectos adversos limitados de la radiación. Existe mayor controversia en los ependimomas supratentoriales por los efectos que podrían darse sobre la cognición. Algunos estudios plantean que el uso de técnicas conformaciones serían mejor toleradas 27.
Actualmente no se ha comprobado que la quimioterapia mejore la sobrevida a cirugía más radioterapia top, sin embargo, esto podría ser materia de futuros estudios cooperativos 28.
Astrocitoma bajo grado cerebelosoEs el tumor más frecuente del sistema nervioso central en la edad pediátrica, corresponde al 10 a 20% de los tumores cerebrales y al 30 a 40% de los tumores de la fosa posterior en los niños. Se presenta desde el primer año de vida hasta la edad adulta, con un peak de incidencia en la mitad de la primera década. Afecta por igual a hombres y mujeres 29,30. En los adultos la mayoría compromete los hemisferios cerebrales, a diferencia de los niños que el 55% de todos los astrocitomas de bajo grado se ubican en la región infratentorial (Figura 7).

El cuadro clínico es de instalación lenta y progresiva dado que son tumores de crecimiento lento, dependerá fundamentalmente de la localización del tumor. Generalmente se presenta como un síndrome de hipertensión intracraneal asociado a un síndrome cerebeloso hemisférico, aunque puede existir también un síndrome cerebeloso de línea media. Ocasionalmente se puede observar compromiso de pares craneales y meningismo. En algunos casos se diagnosticará como hallazgo asintomático al tener neuroimágenes solicitadas por trauma u otros motivos 31.
Es un tumor que en general crece lateralmente, aunque también puede ser vermiano. El tumor hemisférico generalmente es quístico con un nódulo mural, en cambio el tumor vermiano tiende a ser sólido y más frecuente en niños pequeños.
Las neuroimágenes en astrocitomas de bajo grado de la fosa posterior son bastante características, en los astrocitomas grado I son típicas, intensamente homogéneas, bien circunscritas, captan contraste y un edema perilesional mínimo, brillantes en secuencias T1 y T2, quísticas en el cerebelo y con un nódulo mural que capta contraste. En astrocitomas grado II son menos captantes de contraste, aunque suelen haber sobreposición de lesiones grado I y II.
El tratamiento de los astrocitomas de bajo grado se basa en los tres pilares clásicos de la neuro-oncología, cirugía, quimioterapia y radioterapia.
La cirugía es la principal herramienta y su objetivo es la resección lo más completa posible con lo cual es curativa, se obtiene muestra para estudio histológico y molecular, además de resolver la hidrocefalia que muchas veces acompaña a este tipo de tumor. En algunos casos es aceptable una resección parcial o subtotal dada la naturaleza benigna y el lento crecimiento de estas lesiones que incluso en algunos tipos de tumor con la edad se puede detener en forma completa 32–35.
Así, con extirpación total existe un 90 a 100% de sobrevida a 10 años y con extirpación subtotal un 75% de sobreviva a 5 años. En el astrocitoma pilocítico existe un 95% de sobrevida a 5 años y en el astrocitoma difuso un 38% de sobrevida a 5 años. Con la extirpación subtotal se observa un 35% de recurrencia y esta se presenta en un 40% de los casos después de los cinco años del diagnóstico. Esto implica un seguimiento prolongado de este tipo de pacientes.
En pacientes que no es posible una resección completa o que en el seguimiento se demuestra un crecimiento tumoral, la quimioterapia se ha transformado en una opción de tratamiento 31 para lograr un control del crecimiento de los astrocitomas de bajo grado. Hay diferentes esquemas que ofrecen sobrevida libre de enfermedad y sobrevida en general comparables, son relativamente bien toleradas y permiten una buena calidad de vida.
Por último, la radioterapia es la modalidad de tratamiento usada con mayor cautela en la edad pediátrica, la radiación focal es efectiva en la mayoría de los casos de gliomas de bajo grado, sin embargo, existen una cantidad de efectos adversos que es inversamente proporcional a la edad del paciente 37. El déficit neurocognitivo con pérdida de memoria y capacidad de concentración son los efectos más frecuentes, seguidos por vasculopatías, tumores secundarios y endocrinopatías. Dado que la mayoría de los pacientes con gliomas de bajo grado tienen sobrevidas largas los efectos adversos de la radiación son un tema a tener muy en consideración. La radiocirugía también ha sido utilizada como alternativa terapéutica, logrando buen control de la enfermedad, pero los efectos adversos aún están en evaluación.
Del estudio genético de los astrocitomas se concluye que existe un gran número de alteraciones del genoma de significado desconocido. Las alteraciones citogenéticas detectadas mediante técnicas de amplificación y deleción muestran que afectan a prácticamente la totalidad de los cromosomas. A esto hay que añadir las alteraciones debidas a mutaciones puntuales y a cambios epigenéticos como la hipermetilación de genes promotores. Se ha observado que existen unas alteraciones moleculares propias de cada grado histológico tumoral. De forma paralela al proceso de malignización histológica, existe una secuencia de eventos genéticos, que incluyen la amplificación del gen PDGF/R y la mutación del gen TP53 como acontecimientos moleculares iniciales, y la alteración de los genes EGFR y PTEN como alteraciones tardías. La investigación molecular actual ha demostrado que en los astrocitomas hereditarios existen unos genes alterados causantes del desarrollo tumoral, que se conocen con bastante aproximación. Se trata de unas proteínas activadoras de la GTPasa en la neurofibromatosis (17q11 y 22q12), del complejo responsable de las reparaciones de ADN en el síndrome de Turcot (3p21 y 7p22) y de la proteína p53 en el síndrome de Li-Fraumeni (17p13)36.
Tumores de tronco cerebralLos tumores de tronco cerebral corresponden al 10 a 20% del total de tumores intracraneales en niños, de estos un 15 a 20% son astrocitomas de bajo grado que poseen un curso de crecimiento lento y clínicamente se comportan de forma caracteristica, son importantes de diferenciar ya que su tratamiento será muy diferente de los gliomas intrínsecos difusos (GID) que son los más frecuentes 38 (Figuras 8 y 9)


Aproximadamente el 75% corresponden a lesiones difusas intrínsecas del tronco cerebral, que afecta por igual ambos sexos y tienen una edad de presentación más frecuente entre los 7 y 9 años.
Este tipo de tumor es considerado uno de los cánceres más difíciles de tratar en la edad pediátrica, histológicamente corresponden a un heterogéneo grupo de tumores.
Clínicamente se presenta con un cuadro de evolución progresiva en semanas o meses, con compromiso uni o bilateral de pares craneales, compromiso de vías largas piramidales y/o sensitivas y signos cerebelosos. También puede presentarse irritabilidad, letargia y pérdida de peso. La asociación de hidrocefalia es menos frecuente que en los tumores intraventriculares.
Generalmente es de ubicación protuberancial y de tipo difuso, aunque también puede ser focal y tener prolongación exofítica hacia el cuarto ventrículo. Existen diferentes clasificaciones basadas principalmente en las imágenes, que se han ido modificando en relación a los avances de la neuro imagen (Tabla 3).
CLASIFICACIONES DE LOS TUMORES DE TRONCO CEREBRAL SEGÚN DISTINTOS AUTORES
| Epstein et al.39 | Intrínsecos | 1. Difuso 2. Focal 3. Cérvicomedular |
| Exofíticos | 1. Anterolateral en el ángulo pontocerebeloso 2. Posterolateral pontino | |
| Difuso | 1. Citología positiva 2. Mielografía positiva | |
| Epstein et al.40 | Difuso | |
| Focal | ||
| Cérvicomedular | ||
| Stroink et al.41 | Grupo I | Glioma exofítico dorsal |
| Grupo IIa | Tumor intrínseco tronco hipodenso, no capta contraste | |
| Grupo IIb | Tumor intrínseco tronco, capta contraste, exofítico | |
| Grupo III | Tumor focal quístico con captación de contraste | |
| Grupo IV | Focal intrínseco isodenso, capta contraste | |
| Barkovich et al.42 | Localización | Mesencéfalo, protuberancia, bulbo raquídeo |
| Focalidad | Difuso o focal | |
| Dirección y extensión de crecimiento del tumor | ||
| Grado de engrosamiento del tumor | ||
| Crecimiento exofítico | ||
| Hemorragia o necrosis | ||
| Evidencia de hidrocefalia | ||
| Albright43 | Difuso | |
| Focal | Mesencéfalo, protuberancia intrínseca, exofítico dorsal, bulbo raquídeo | |
| Fischbein et al.44 | Mesencéfalo | Difuso Focal Tectal |
| Protuberancia | Difuso Focal | |
| Bulboraquídeo | Difuso Focal Dorsal exofítico | |
| Rubin et al.45 | Cérvicomedular | |
| Exofítico | ||
| Quístico | ||
| Focal | ||
| Difuso | ||
| Choux et al.46 | Tipo I – difuso | |
| Tipo II – intrínseco, focal | ||
| Tipo III – exofítico, focal | ||
| Tipo IV – cérvicomedular | ||
Tabla traducida de: Surgery of the pons, Pablo Recinos, Violette Renard, George Jallo, Posterior Fossa Tumors in Children, 559 DOI 10.1007/978-3-319-11274-9_14, ¿ Springer International Publishing Switzerland 2015:M.M. Ozek et al. (eds.).
El diagnóstico puede realizarse por biopsia a cielo abierto o estereotáxica, pero es discutible y actualmente en muchos casos no se realiza debido a que la resonancia magnética entrega información con alto grado de certeza y permite iniciar más precozmente el tratamiento.
Del punto de vista terapéutico es un tumor de indicación quirúrgica limitada a lesiones de características intrínsecas focales o exofítico y sea abordado como tumor intraventricular o focal. La quimioterapia actualmente no tiene utilidad clara, por lo cual la terapia más utilizada es la radioterapia craneoespinal a la brevedad, lo que permite controlar su crecimiento y ofrecer una mejor calidad de vida a pesar del ominoso pronóstico de sobrevida.
La radioterapia y un buen cuidado de soporte pueden llevar a sobrevidas de 8 a 11 meses, no hay estudios de quimioterapia que demuestren mejorías significativas en la sobrevida, por lo tanto, ninguna medicación se considera un estándar de tratamiento 47.
El avance en el conocimiento de los gliomas difusos intrínsecos del tronco se ha visto limitado por la escasez de muestras histológicas para estudios moleculares. La evolución de las imágenes limitó de forma importante la obtención de muestras de tejido para diagnóstico, por eso hoy en día hay una tendencia al cambio de estrategia y se recomienda la realización de biopsias diagnósticas, en especial cuando hay imágenes atípicas. También se estimula la donación de tejido tumoral en pacientes que fallecen. El estudio post mortem ha entregado mucha información molecular y biológica de los gliomas difusos del tronco, estudios basados en secuenciación del genoma y de todo el exoma han revelado que el 80% de los gliomas difusos intrínsecos del tronco los glioblastomas multiformes del tálamo poseen mutaciones de la HIST1H3B, HIST1H3C y H3F3A. Esto ha llevado a proponer una clasificación basada en las mutaciones histona H3, MYCIN y grupos silentes 48.
Los GID frecuentemente contienen gruesas aberraciones cromosómicas con ganancia en 1q, 2q, 7p, 7q, 8q, and 9q y pérdida 4q,16q, 17p y 20p 49. En fin, hay numerosas líneas de investigación que se espera puedan dar nuevas direcciones en identificar agentes molecularmente dirigidos que puedan actuar sinérgicamente, lo cual permitirá mejorar las opciones terapéuticas en los GID.
En suma, los tumores de tronco cerebral en la edad pediátrica son un grupo heterogéneo y su comportamiento dependerá de su ubicación, focal o difuso, histología y características genéticas. En los casos en que el tumor es focal o exofítico, la cirugía sigue siendo el tratamiento de elección siendo un gran desafío desde el punto de vista quirúrgico por lo elocuente de la zona, en los casos difusos la radioterapia es la mejor alternativa. La quimioterapia tiene un papel limitado y sin duda los avances en la biología molecular de estos tumores permitirá diseñar tratamientos específicos para cada individuo.
CONCLUSIONESLos tumores del sistema nervioso central de la fosa posterior en la edad pediátrica son una patología frecuente, predominando en el grupo etáreo de los 4-9 años. Son un grupo muy heterogéneo de tumores en su presentación clínica, localización, neuroimagen, histología, comportamiento biológico y tratamiento. Los avances de la medicina en este último tiempo se han enfocado en lograr una mejor caracterización molecular de cada uno de estos tumores, esto con el objetivo de lograr diseñar tratamientos acordes a cada uno de éstos para que sean efectivos y con mínimos efectos secundarios. Se ha avanzado mucho en algunos casos como el meduloblastoma en que se han definido cuatro grupos desde el punto de vista molecular, que se sabe de antemano tendrán una conducta muy diferente y por lo tanto, requieren un manejo hecho “a la medida”. En los tumores de tronco y en especial en los gliomas intrínsecos difusos, de tan mal pronóstico actual, hay un muy interesante desarrollo que sin duda permitirá en un futuro próximo diseñar fármacos que actuarán de manera sinérgica y junto a la radioterapia permitirán mejorar la sobrevida de este catastrófico tumor.
Por último, hay que insistir que para ser exitosos en el manejo de estos tumores el trabajo cooperativo es esencial para la investigación y desarrollo de los estudios moleculares que permitirán conocer mejor la naturaleza de cada paciente y su tumor, permitiendo así diseñar mejores tratamientos. A nivel de cada paciente es fundamental el trabajo en equipo con un grupo multidisciplinario que debe incluir: pediatras, neurocirujanos, neurorradiólogos, neuropatólogos, oncólogos, radioterapeutas, neurólogos, psiquiatras, enfermeras, nutricionistas, kinesiólogos y psicólogos. Cada paciente, familia y su tumor, son un ente único que requiere un manejo individualizado para ser exitoso.
El autor declara no tener conflictos de interés, en relación a este artículo.