




Los tumores malignos en pediatría representan sólo el 2% de los casos de cáncer, sin embargo, las neoplasias son en la actualidad la segunda causa de muerte en niños mayores de 1 año de edad. Cada año se diagnostican aproximadamente 130 nuevos casos de cáncer por millón de niños. La leucemia es el más frecuente de los cánceres pediátricos, seguidos por tumores de cerebro, linfomas, neuroblastomas, sarcomas, tumores de Wilms y tumor de células germinales. La probabilidad de sobrevivir a una enfermedad maligna ha mejorado desde que en 1950 se reportaron las primeras remisiones en leucemia linfocítica aguda. Actualmente, debido al desarrollo de la quimioterapia, mejora en los métodos diagnósticos y el manejo multidisciplinario de los pacientes, el porcentaje de curación es cercano a un 75%. A pesar de estos avances, es aún necesario una mejoría de los resultados en el cáncer pediátrico, que depende del diagnóstico temprano de la enfermedad.
Cancer in children represents only 2% of all malignancies, nevertheless, after trauma, it is the second most common cause of death in children older than 1 year. Each year approximately 130 new cases of cancer are diagnosed per million children. Leukemia is the most common form of cancer in children, followed by brain tumor, lymphoma, neuroblastoma, sarcoma, Wilm's tumor and germ cell tumor. The probability of surviving childhood malignancies has improved since the first remissions in acute lymphoblastic leukemia were reported in 1950s. At the moment due to the development of chemotherapy regimens, improvement in diagnostic methods and multidisciplinary approach of our patients, the survival is close to 75%. Therefore, there is still a need for significant improvement in the results of childhood cancer.
Los tumores malignos en pacientes pediátricos son infrecuentes, representan sólo el 2% de todos los casos de cáncer, la incidencia estimada es de 12–14 casos de cáncer por 100.000 niños menores de 15 años, en Chile, se estiman 450 a 540 casos nuevos por año (1), sin embargo, las neoplasias son en la actualidad la segunda causa de muerte en pediatría, luego de los accidentes en niños mayores de 1 año de edad, siendo la tasa de mortalidad de un 3,7 por 100.000 niños (1, 2).
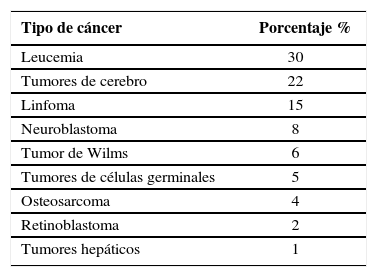
La leucemia es el más frecuente de los cánceres pediátricos, seguidos por los tumores de cerebro (TC), linfomas, neuroblastomas (NBL), sarcoma de partes blandas (SPB), tumores de Wilms (TW), tumor de células germinales (TCG) y retinoblastoma (Tabla 1). La distribución cambia según la edad, en pacientes entre 15 y 18 años, la enfermedad de Hodgkin y tumores de células germinales son los tumores más frecuentes, osteosarcomas (OSC), tumores de Ewing (TE) y melanoma también incrementan en este grupo etario.
Frecuencia De Tumores Malignos De La Infancia
| Tipo de cáncer | Porcentaje % |
|---|---|
| Leucemia | 30 |
| Tumores de cerebro | 22 |
| Linfoma | 15 |
| Neuroblastoma | 8 |
| Tumor de Wilms | 6 |
| Tumores de células germinales | 5 |
| Osteosarcoma | 4 |
| Retinoblastoma | 2 |
| Tumores hepáticos | 1 |
Krasin M, Davidoff A. Principles of pediatric Oncology, Genetics of cancer, and radiation Therapy. En Grosfeld J, Fonkalsrud E., Corn A. Pediatric Surgery. 6taEdicion. Philadelphia. mosby. 2006:412.
En general, la incidencia de los tumores en la infancia es mayor en el primer año de edad, con un segundo incremento a los 2 a 3 años declinando luego hasta los 9 años de edad, aumentando nuevamente en la adolescencia, la distribución varía según la histología, género y raza (3). La probabilidad de sobrevivir a una enfermedad maligna ha mejorado desde que en 1950, Farber indujo las primeras remisiones enleucemia linfocítica aguda (LLA) (4). Actualmente el porcentaje de curación ha incrementado a cerca de un 75%, esto es debido al desarrollo de quimioterapia, a la buena respuesta que inicialmente tienen los tumores malignos pediátricos y al manejo multidisciplinario. A pesar de estos avances queda un largo camino por recorrer para aumentar la sobrevida de tumores malignos que aún presentan pobre sobrevida como neuroblastomas o rabdomiosarcomas (RMS).
Desde 1987, el tratamiento del cáncer pediátrico en Chile se efectúa de acuerdo a protocolos nacionales, enmarcados dentro del Programa Nacional de Drogas Antineoplásicas (PINDA) del Ministerio de Salud. Esto mejoró la sobrevida de algunos cánceres ostensiblemente, ejemplo claro es la leucemia linfoblástica aguda que mejoró su sobrevida libre de enfermedad desde 51% a 91% a 5 años (5).
El diagnóstico precoz en el cáncer pediátrico juega un rol preponderante en el incremento de la sobrevida, es por esto que es fundamental que el pediatra general conozca y maneje los aspectos básicos de las patologías frecuentes en oncología infantil, para así realizar una derivación precoz y oportuna. El objetivo de este artículo es entregar conceptos generales del cáncer del niño, destinado a los médicos pediatras generales. Se detallan algunos aspectos epidemiológicos, diagnósticos y terapéuticos de la patología quirúrgica oncológica.
Biología molecular del cáncerDurante el desarrollo normal de las células, éstas se transforman llegando a tener funciones altamente específicas. El desarrollo y renovación celular son una serie de procesos regulados que incluyen proliferación, diferenciación y muerte programada (apoptosis). El cáncer es una enfermedad cuyo desarrollo está determinado por una serie de mutaciones génicas, asociadas a factores hereditarios y del ambiente. Estos cambios resultan en células con un fenotipo que favorece su sobrevida comparada con células normales, produciendo el crecimiento desregulado que caracteriza a las células neoplásicas. La oncogénesis refleja la acumulación de una excesiva proliferación celular y la disminución de la apoptosis. Las células cancerígenas presentan DNA con puntos de mutación, inserciones virales y/o amplificaciones o delecciones cromosómicas. Cada una de estas aberraciones puede alterar el proceso normal de crecimiento y diferenciación celular. Anormalidades genéticas asociadas con el cáncer pueden ser detectadas en cualquier célula del cuerpo y/o en las células tumorales (6).
Tratamiento del cáncer en la infanciaEl tratamiento de las patologías oncológicas es multimodal, dependiendo de la histología tumoral. Los diferentes componentes terapéuticos: quimioterapia, inmunoterapia, trasplante de médula ósea, radioterapia y cirugía, juegan roles diferenciales con el objetivo de obtener sobrevida libre de enfermedad, con la menor cantidad de afectos adversos posibles.
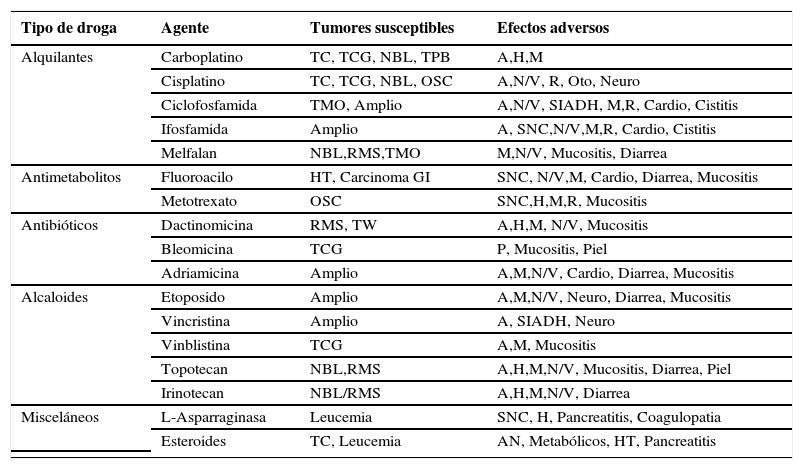
QuimioterapiaActualmente la quimioterapia es parte integral del tratamiento de casi todos los cánceres en la infancia. El propósito de esta terapia es provocar la muerte de las células tumorales minimizando el daño a las células normales (7). Luego de un promisorio comienzo en la utilización de esta terapia contra los tumores pediátricos, se ha evidenciado últimamente que el éxito de la terapia se ve disminuido por la aparición de resistencia hacia diferentes agentes y la generación de toxicidad a tejidos normales. Sin embargo su uso mantiene un importante rol como adyuvante en la reducción tumoral en enfermedades localizadas y como el principal tratamiento de tumores diseminados. Los agentes quimioterápicos más comunes se muestran en la Tabla 2(8).
Agentes Comunes En Quimioterapia
| Tipo de droga | Agente | Tumores susceptibles | Efectos adversos |
|---|---|---|---|
| Alquilantes | Carboplatino | TC, TCG, NBL, TPB | A,H,M |
| Cisplatino | TC, TCG, NBL, OSC | A,N/V, R, Oto, Neuro | |
| Ciclofosfamida | TMO, Amplio | A,N/V, SIADH, M,R, Cardio, Cistitis | |
| Ifosfamida | Amplio | A, SNC,N/V,M,R, Cardio, Cistitis | |
| Melfalan | NBL,RMS,TMO | M,N/V, Mucositis, Diarrea | |
| Antimetabolitos | Fluoroacilo | HT, Carcinoma GI | SNC, N/V,M, Cardio, Diarrea, Mucositis |
| Metotrexato | OSC | SNC,H,M,R, Mucositis | |
| Antibióticos | Dactinomicina | RMS, TW | A,H,M, N/V, Mucositis |
| Bleomicina | TCG | P, Mucositis, Piel | |
| Adriamicina | Amplio | A,M,N/V, Cardio, Diarrea, Mucositis | |
| Alcaloides | Etoposido | Amplio | A,M,N/V, Neuro, Diarrea, Mucositis |
| Vincristina | Amplio | A, SIADH, Neuro | |
| Vinblistina | TCG | A,M, Mucositis | |
| Topotecan | NBL,RMS | A,H,M,N/V, Mucositis, Diarrea, Piel | |
| Irinotecan | NBL/RMS | A,H,M,N/V, Diarrea | |
| Misceláneos | L-Asparraginasa | Leucemia | SNC, H, Pancreatitis, Coagulopatia |
| Esteroides | TC, Leucemia | AN, Metabólicos, HT, Pancreatitis |
A: Alopecia; SNC: Sistema Nervioso Central; H: Hepatotoxicidad; M: Mielosupresor; N/V: Náusea/Vómito; P: Pulmonar; R: Renal; SIADH: Síndrome de secreción inadecuada de ADH; HT: Hipertensión; TMO: Trasplante de Médula Ósea; TC: Tumor cerebral; TCG: Tumor de células germinales; NBL: Neuroblastoma; OSC: Osteosarcoma; RMS: Rabdomiosarcoma; TPB: Tumor de partes blandas; TW: Tumor de Wilms.
Es una de las tres modalidades terapéuticas principales en el tratamiento del cáncer. Es utilizada como terapia única o en combinación con quimioterapia y/o cirugía. La radioterapia puede ser utilizada preoperatoria para disminución de masa tumoral o post-operatoria en casos en que la resección quirúrgica no fue completa o en resecciones marginales, ésta es la indicación más frecuente en casos pediátricos. La radiación puede ser entregada en forma externa, donde se define el área a irradiar con diferentes imágenes, o en forma local insertando catéteres en la zona de resección tumoral, lo que permite radiación directa de tumor residual disminuyendo las complicaciones de la radiación a tejidos adyacentes normales. Tumores comúnmente irradiados son sarcomas de partes blandas, RMS, Sarcoma de Ewing, tumores de Wilms y en el tratamiento de leucemias y linfomas (9).
CirugíaCon el avance de las otras modalidades terapéuticas como la quimioterapia y radioterapia, la cirugía se ha convertido en un elemento más del armamento terapéutico en la cura del cáncer, sin embargo en ciertos tumores la extirpación quirúrgica sigue siendo imprescindible. El tratamiento quirúrgico está inmerso en un tratamiento multimodal que varía según diversos protocolos (10). El progreso de las técnicas quirúrgicas y la implementación de la cirugía mínimamente invasiva, hacen que la cirugía oncológica sea uno de los campos más dinámicos de la práctica quirúrgica pediátrica (11).
Rol de la cirugía en el cáncer pediátricoRol diagnósticoEn el pasado, el diagnóstico definitivo era hecho al momento de la resección quirúrgica del tumor primario. Actualmente, con el avance de la terapia sistémica, la mayoría de los cánceres infantiles son tratados preoperatoriamente, por tanto muchos pacientes requieren diagnóstico con biopsias percutáneas o incisionales. En la actualidad, con el conocimiento de los cambios moleculares genéticos asociados a estas patologías, el diagnóstico y estadío es logrado con especímenes mucho más pequeños que los requeridos previamente. Esto ha llevado al avance en las técnicas de biopsia para lograr la toma de la muestra sin producir o limitando la morbilidad del procedimiento. Actualmente se utiliza cuando es posible biopsia percutánea o el acceso mínimamente invasivo para diagnóstico de un tumor primario y en sospecha de metástasis o recurrencia.
Rol terapéuticoLa cirugía está presente en diversos niveles durante el manejo del cáncer infantil: extirpación, manejo de metástasis, implantación de accesos vasculares y en el tratamiento de las complicaciones asociadas al tumor.
Extirpación tumoral: Los tumores que se benefician de la extirpación quirúrgica se resumen en la Tabla 3. Dependiendo del estadío tumoral, la cirugía tiene un papel más o menos relevante en diversas histologías, por ejemplo en el Neuroblastoma la cirugía es curativa por sí sola en tumores localizados de histología favorable (estadíos INSS 1 o 2), en estadíos más avanzados la cirugía resectiva mejora la sobrevida cuando se asocia a terapia sistémica (12). En el tumor de Wilms la extirpación completa del tumor es imprescindible para la curación de la enfermedad en todos sus estadíos complementada con quimioterapia, y radioterapia en casos específicos (13). En tumores de origen mesenquimático y RMS localizados, la resección es suficiente en el manejo de la enfermedad, en todos los demás estadíos y localizaciones, la quimioterapia asociada o no a radioterapia deben ser utilizados, realizando extirpación quirúrgica cuando es posible (14). En hepatoblastoma la resección también es imprescindible en el manejo, antecedido por quimioterapia para reducción tumoral en la mayoría de los casos. En casos de hepatoblastomas en los que la resección quirúrgica con márgenes oncológicos no es posible, la extirpación tumoral completa se logra sustituyendo el órgano con un trasplante hepático, lo que ha logrado aumentar la sobrevida de pacientes antes considerados irresecables (15). En tumores óseos de extremidades y torácicos, la resección quirúrgica con márgenes amplios es fundamental, pero el tratamiento requiere la utilización de quimioterapia y en ocasiones radioterapia para mejorar la sobrevida. La cirugía en suma es primordial para la curación de diversos tumores, asociada o no terapia sistémica para lograr reducción tumoral.
Tumores Que Se Benefician De Resección Quirúrgica
| Neuroblastoma Estadío 1 y 2 |
| Tumor de Wilms* |
| Neuroblastoma Estadío 3 y 4* |
| Tumor mesenquimal maligno |
| Hepatoblastoma* |
| Tumores óseos |
| Tumor de células germinales |
| Tumor suprarrenal |
| Tumor tiroideo |
Extirpación de las metástasis: En tumores en los cuales la terapia sistémica tiene un rol limitado en el manejo de la enfermedad por mala respuesta a quimioterapia, la resección de metástasis se transforma en un elemento primordial en la mejora de la sobrevida del paciente, esto es claro en tumores de origen mesenquimático, tumores óseos (osteosarcoma y Ewing), hepatoblastoma y tumor de Wilms. El manejo quirúrgico asociado a quimioterapia y radioterapia, en algunos casos puede ser curativo o prolongar sustancialmente la sobrevida.
Acceso vascular: El desarrollo de accesos vasculares permanentes ha facilitado el manejo de pacientes oncológicos disminuyendo el riesgo de extravasaciones del agente quimioterápico y el trauma de punciones venosas repetidas durante el manejo de la enfermedad. Actualmente la mayoría de los pacientes oncológicos recibe tratamiento quimioterápicos a través de un acceso vascular implantado al diagnóstico de la enfermedad.
Manejo de complicaciones derivadas del tumor o su tratamiento: El cirujano oncólogo pediatra juega un rol importante en el manejo de secuelas derivadas del tumor o su tratamiento, es frecuente la necesidad de soporte nutricional a través de gastrostomías, creación de traqueostomía en casos de compromiso de vía aérea, creación de derivaciones urinarias en el manejo de tumores pélvicos, manejo de escoliosis luego de resecciones masivas torácicas, manejo de complicaciones infecciosas y prevención de secuelas como es la movilización de los ovarios para preservar en lo posible las células germinales en los casos que se requiere terapia pélvica como en el tratamiento del linfoma y sarcomas pélvicos.
Tumores sólidos de manejo quirúrgico en pediatríaCabeza y cuelloDiversos tumores pueden afectar esta área anatómica, como rabdomio-sarcomas, linfomas, carcinoma de células escamosas, melanomas, sarcomas de partes blandas y tumores de nervio periférico la mayoría tratados con terapia sistémica, pues la resección quirúrgica es mutilante sin mejorar sobrevida. Los tumores quirúrgicos más frecuentes son los tumores cerebrales y tiroideos.
Tumores cerebrales: son los tumores sólidos más frecuentes en la oncología infantil, con una incidencia de 3.1 por 100.000 niños. La cirugía se realiza para definir el diagnóstico histológico y para restablecer el flujo de líquido cefalorraquídeo cuando es requerido. Para la mayoría de los tumores de sistema nervioso central la resección tumoral, aunque no sea completa, es indicada, pues mejora el pronóstico cuando se asocia a quimioterapia y radioterapia (16).
Tumores tiroideos: Los tumores malignos tiroideos son infrecuentes, un 10% de los cánceres tiroideos ocurren en pacientes menores de 21 años de edad. Representan un 3% de los tumores sólidos de la infancia, el peak de incidencia ocurre entre los 10 y 18 años. Su incidencia aumenta en pacientes que han recibido radioterapia previamente (17). Los tumores tiroideos bien diferenciados, usualmente carcinoma papilar, son los más frecuentes. Los carcinomas medulares, foliculares y mixtos son menos frecuentes. El diagnóstico incluye cintigrafía tiroidea y ultra-sonografía. La aspiración con aguja fina para diagnóstico puede ser utilizada como alternativa a la biopsia incisional. El tratamiento quirúrgico comprende tiroidectomía total o parcial con ablación con I131 y disección ganglionar cervical cuando es indicada. Aproximadamente el 5% de los cánceres tiroideos son carcinomas medulares, los que se generan en las células parafoliculares, la resección quirúrgica es el único tratamiento de este tumor. El carcinoma medular puede ser esporádico o asociarse a tumores familiares asociados a síndromes MEN 2A, MEN 2B o FMTC. En niños de familias con MEN 2, se realiza detección del proto-oncogen RET, responsable de la enfermedad, si está presente, se sugiere realizar tiroidectomía total profiláctica a la edad de cinco años. Aproximadamente en un 80% de estas tiroides se encuentran focos de tumor medular (18).
TóraxLos tumores torácicos se deben dividir en tumores de pared torácica, pulmón y mediastino;
Tumores de pared torácica son infrecuentes, siendo mayoritariamente rabdomiosarcoma y tumores de Ewing (también denominados PNET o Askin tumor) derivados de la pared costal, así como sarcomas de partes blandas y osteosarcomas, otros tumores que afectan la pared torácica son tumores benignos como osteocondromas, quistes aneurismáticos (19). La resección quirúrgica es fundamental en todas las histologías, requiriendo márgenes quirúrgicos extensos, lo que lleva a importantes secuelas a largo plazo, especialmente escoliosis.
Tumores malignos pulmonares primarios en la infancia son muy infrecuentes, siendo el adenoma bronquial el más frecuente; sigue en frecuencia el carcinoma broncogénico, que se presenta mayoritariamente en pacientes con malformaciones adenomatoideas quísticas (MAQ) o quistes broncogénicos; otra neoplasia asociada a patología congénita del pulmón es el blastoma pleuropulmonar, asociado a secuestros intralobares y MAQ (20). La mayoría de los tumores pulmonares en pediatría corresponden a metástasis. El tumor metastásico más común es el tumor de Wilms. Otros tumores que presentan diseminación pulmonar son los sarcomas óseos y de partes blandas, especialmente rabdomiosarcoma, tumores tiroideos, hepáticos, melanoma y teratocarcinoma. El manejo de las metástasis pulmonares depende de la histología, prefiriendo resección en los tumores de Wilms, osteosarcoma, hepatoblastoma y tumores mesenquimales (21).
Tumores mediastínicos: El 40% de las masas mediastínicas en la infancia corresponden a tumores malignos, la mayoría son encontradas incidentalmente. La localización anatómica de la masa sugiere el origen de la lesión, tumores de mediastino posterior son frecuentemente de origen neurogénicos como neuroblastomas o ganglioneuromas; tumores de mediastino anterior son mayoritariamente linfomas y en niños pequeños tumores de células germinales como teratomas. Dependiendo de las características en imágenes del tumor y la presencia de otras lesiones sistémicas, se debe decidir si la resección primaria del tumor es indicada (posible teratoma o tumores neurogénicos) o es apropiada una biopsia. En tumores mediastínicos el acceso toracoscópico para biopsia o resección es una de las alternativas a considerar. En artículos previamente publicados este acceso tiene una efectividad del 93% en la determinación del diagnóstico (22).
AbdomenLos tumores abdominales más frecuentes en pediatría son el neuroblastoma, tumor de Wilms, tumor de células germinales y hepatoblastoma.
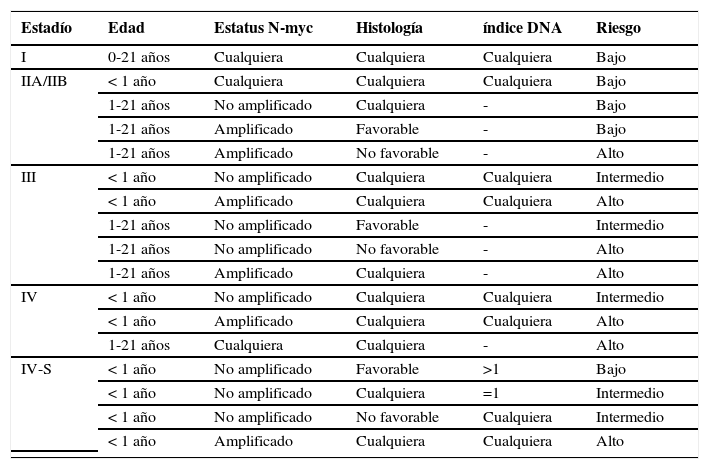
Neuroblastoma: Esta neoplasia se origina en las células de la cresta neural y a lo largo de la cadena simpática desde el cuello a la pelvis. Su curso clínico es variable, se han reportado regresiones espontáneas y maduración tumoral, sin embargo la enfermedad es progresiva en la mayoría de los casos. A diferencia de otros tumores la sobrevida en tumores metastásico no se ha logrado mejorar aún con terapia agresiva multimodal. El neuroblastoma corresponde a un 10% del cáncer en la infancia y produce el 15% de la mortalidad por cáncer. Es asociado a la presencia de síndrome de Beckwith-Wiedemann, neurofibromatosis, enfermedad de Hirschsprung y síndrome fetal alcohólico. El neuroblastoma tiene manifestaciones clínicas diversas dependiendo de la localización, presencia de metástasis y la producción de ciertos metabolitos (12). Cincuenta a 75% se presentan como masa abdominal. Síntomas sistémicos incluyen baja de peso, fiebre, anemia y distensión abdominal. La producción excesiva de catecolaminas puede producir flushing, irritabilidad y temblor. La presencia de ataxia y opsomioclonus ha sido observada en neuroblastomas. La presencia de “ojos de mapache” clásica en la presentación clínica del neuroblastoma es producto de metástasis óseas a la base de cráneo. El diagnóstico se realiza a través de variadas imágenes (Tomografía axial computada (TAC), estudios isotópicos con MIBG-I123 y resonancia magnética), determinación sérica y urinaria de catecolaminas y estudios histológicos y genéticos de tejido tumoral obtenido de medula ósea en caso de tumor metastásico o del tumor primario en casos de tumor localizado. El tratamiento del neuroblastoma es basado en el “Riesgo” (Bajo, Intermedio o Alto; Tabla 4), este está definido según estadio tumoral, edad, presencia de amplificación del gen N-myc en el tejido tumoral (23), ploidia del DNA en tejido tumoral e histología según los criterios de Shimada, estos factores definirán la necesidad de resección quirúrgica aislada, o su asociación a quimioterapia más agresiva dependiendo del riesgo, con la utilización de radioterapia, ablación de médula y trasplante de médula ósea en paciente con alto riesgo sin respuesta a terapia habitual (24).
Grupos de Riesgo en Neuroblastoma
| Estadío | Edad | Estatus N-myc | Histología | índice DNA | Riesgo |
|---|---|---|---|---|---|
| I | 0-21 años | Cualquiera | Cualquiera | Cualquiera | Bajo |
| IIA/IIB | < 1 año | Cualquiera | Cualquiera | Cualquiera | Bajo |
| 1-21 años | No amplificado | Cualquiera | - | Bajo | |
| 1-21 años | Amplificado | Favorable | - | Bajo | |
| 1-21 años | Amplificado | No favorable | - | Alto | |
| III | < 1 año | No amplificado | Cualquiera | Cualquiera | Intermedio |
| < 1 año | Amplificado | Cualquiera | Cualquiera | Alto | |
| 1-21 años | No amplificado | Favorable | - | Intermedio | |
| 1-21 años | No amplificado | No favorable | - | Alto | |
| 1-21 años | Amplificado | Cualquiera | - | Alto | |
| IV | < 1 año | No amplificado | Cualquiera | Cualquiera | Intermedio |
| < 1 año | Amplificado | Cualquiera | Cualquiera | Alto | |
| 1-21 años | Cualquiera | Cualquiera | - | Alto | |
| IV-S | < 1 año | No amplificado | Favorable | >1 | Bajo |
| < 1 año | No amplificado | Cualquiera | =1 | Intermedio | |
| < 1 año | No amplificado | No favorable | Cualquiera | Intermedio | |
| < 1 año | Amplificado | Cualquiera | Cualquiera | Alto |
N-myc: no amplificado = 1 copia, amplificado >1 copia. Indice DNA: aneuploidia >1; diploidia =1
Gosfeld J. Neuroblastoma. En Grosfeld J, O'Neill J, Fonkalsrud E. Pediatric Surgery. 6taEdición. Philadelphia. Mosby. 2006:476.
Tumor de Wilms: El tumor de Wilms afecta a 1 de cada 10.000 niños menores de 15 años, representando el 6% de los cánceres infantiles. La edad promedio de presentación es 38 meses. Pacientes con tumores bilaterales, aniridia, síndrome de Beckwith-Wiedeman (B-W), criptorquidea o hipospadias se diagnostican en forma más precoz (17 a 27 meses de edad). El tumor de Wilms tiene asociación con diferentes anomalías congénitas como la hemihipertrofia, síndrome de B-W y síndrome de WAGR (Wilms, aniridia, anomalías genitourinarias y retardo mental). El síndrome Denish-Drash ha sido asociado a mutaciones del gen WT1, y presentan alto riesgo de presentar tumor de Wilms. Existen también casos de TW familiares, con transmisión hereditaria dominante de penetrancia variable asociadas a los genes FWT1 y FWT2 (25). La presentación clínica habitual es con la presencia de una masa abdominal indolora, hematuria y en raros casos dolor abdominal por ruptura tumoral. El diagnóstico, se realiza a través de estudios por imágenes, se utiliza TAC en la sospecha de TW para evaluación del riñón afectado y evaluación del riñón contralateral, el doppler Ultrasonido se utiliza para detección de compromiso de las venas renal y cava. Es controversial si una radiografía de tórax basta para la evaluación de metástasis pulmonares, o si la TAC torácica es necesaria para su detección. El screening con ultrasonido se realiza cada 4 meses en pacientes con síndromes genéticos que incrementan la incidencia de TW. El tratamiento del TW es diferente según el protocolo aplicado, en general en Europa los pacientes reciben quimioterapia preoperatoria, luego resección y quimioterapia postoperatoria dependiendo de la histología al momento de la resección asociado a radioterapia en casos de anaplasia difusa (26). En Norteamérica se realiza resección quirúrgica cada vez que ésta es posible (excepto en estadío V (TW bilateral) y tumores irresecables) y luego el paciente es tratado con quimioterapia adyuvante que varía según el estadío, edad del paciente y la histología, en estadío 1 (tumor localizado en el riñón, resecado en forma completa) con histología favorable y anaplasia focal o difusa y en estadío II (tumor completamente resecado con extensión regional, previa biopsia o ruptura pre o intraoperatoria confinada al flanco renal) con histología favorable son tratados con Vincristina y Dactinomicina por un total de 18 semanas; pacientes con anaplasia focal, estadío II o III (resección incompleta, contaminación peritoneal, implantes peritoneales o linfonodos positivos), estadío III de favorable histología y estadío IV (Metástasis hematógenas) y estadío V de favorable histología o anaplasia focal se tratan con Vincristina, Dactinomicina y Doxorrubicina, asociado a radioterapia abdominal; pacientes con anaplasia difusa, estadíos II, III, IV reciben Ciclofosfamida, Doxorrubicina, Etoposido y Vincristina asociado a irradiación abdominal. Con este protocolo la sobrevida libre de enfermedad a 2 años es 90% (13, 26).
Tumor de células germinales: Es un grupo heterogéneo de neoplasias, comprende 7 grupos histológicos: disgerminoma, tumor del saco vitelino, carcinoma embrionario, poliembrioma, coriocarcinoma, teratoma y tumor de células germinales mixtos. Presentan una incidencia de 2.4 casos/1.000.000 niños, representando el 1% de los cánceres pediátricos. Estas neoplasias tienen dos presentaciones gonadal y extragonadal, la presentación clínica y las características histopatológicas varían dependiendo de la localización. Los tumores de células germinales provienen de las células germinales primordiales, las que migran en el periodo embrionario a lo largo de la línea media para llegar a conformar el tejido gonadal, si este proceso es alterado, las células germinales se depositan a lo largo del trayecto de migración, por este motivo este tipo de tumores pueden ser encontrados en el área sacrococcígea, retroperitoneo, mediastino, cuello, cerebro y en ovario o testículo. La transformación maligna puede ocurrir en cualquiera de estos sitios anatómicos. La mayoría de estos tumores, tienen marcadores séricos específicos lo que es utilizado en su identificación histológica, en evaluación de la respuesta a terapia y como screening de posibles recurrencias. Los marcadores biológicos son principalmente alfa feto proteína (AFP), elevada en casos de tumores de saco vitelino y carcinoma embrionario y sub-unidad B de la gonadotrofina coriónica humana (b-HCG), elevada en pacientes con coriocarcinoma, seminoma o disgerminoma y ocasionalmente en carcinoma embrionario. El manejo terapéutico óptimo se logra con resección quirúrgica completa de la lesión, evaluación cuidadosa de la histología y la utilización selectiva de quimioterapia. La sobrevida actual en tumores de bajo estadío en localización extra gonadal y tumores gonadales de bajo o alto estadíos, es de 90% a 100%. La sobrevida de tumores extra gonadales de estadíos altos es de 75% (27).
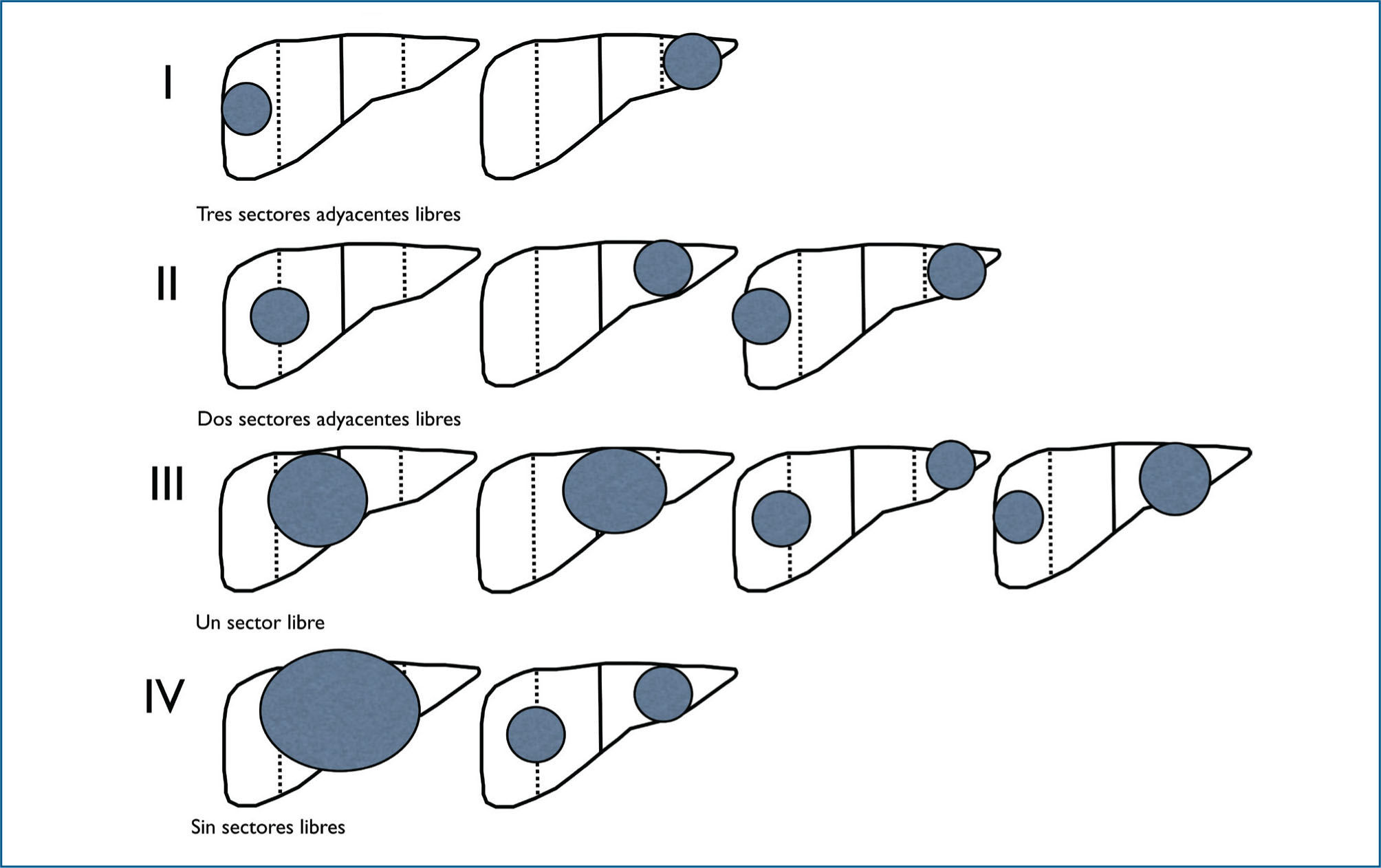
Hepatoblastoma: La incidencia del hepatoblastoma es 1,2 casos/1.000.000 niños, representa el 1% de los cánceres en pediatría. Ocurre principalmente en hombres, tiene un peak de presentación entre los 6 meses y 3 años de edad. El hepatoblastoma está asociado a síndromes genéticos como el síndrome de B-W, adenomatosis poliposa familiar, hemihipertrofia y prematurez. La mayoría de los pacientes se presentan con masa abdominal indolora, se puede asociar a baja de peso, anorexia y fatiga, ante estos hallazgos se debe realizar perfil bioquímico completo, hemograma y niveles de AFP séricos junto con scanner abdominal y torácico. El diagnóstico definitivo se realiza con biopsia tumoral, que se puede realizar con acceso laparoscópico, cirugía abierta convencional o percutáneo. El diagnóstico deferencial de una masa hepática son el hepatocarcinoma (presente en pacientes mayores de 5 años, usualmente adolecentes), sarcoma embrionario y rabdomiosarcoma (usualmente entre los 5 y 10 años de edad), otros tumores presentes en el hígado son angiosarcoma, teratoma inmaduro, tumor rabdoide, hamartomas y coriocarcinomas, la enfermedad metastásica es poco frecuente en la patología pediátrica. El hepatoblastoma se clasifica histológicamente en 2 tipos principales: tumores epiteliales (fetal, embrional, macrotrabecular y tumor indiferenciado de células pequeñas) y tumores mixtos: epitelial y mesenquimatico, esta diferenciación no determina valor pronóstico, excepto en tumores de tipo fetal que se asocian a mejor sobrevida y el tumor indiferenciado de células pequeñas que confiere un mal pronóstico. Existen actualmente diferentes métodos para evaluar estadío tumoral, Estados Unidos utiliza un esquema basado en los hallazgos intraoperatorios, tumores resecados completamente (Stage 1), enfermedad residual microscópica (Stage II), enfermedad residual macroscópica (Stage III) y enfermedad extra hepática o metastásica (Stage IV), esta clasificación es útil para definir pronóstico postoperatorio, pero no provee información de la extensión de la enfermedad preoperatoria. En la evaluación de resecabilidad y la respuesta a quimioterapia es útil la clasificación utilizada por la Sociedad Internacional de Oncología Pediátrica (SIOP), este grupo desarrollo el PRETEXT (extensión tumoral pre-tratamiento), clasificación basada en los hallazgos radiológicos, y describe el número y localización de las lesiones, y además toma en cuenta la invasión de las venas supra hepáticas y porta, así como enfermedad extra hepática y metastásica. El hígado es dividido en diferentes sectores que definen la extensión tumoral (Figura 1) (28). El tratamiento del hepatoblastoma depende del protocolo utilizado, en general se trata con quimioterapia preoperatoria, reevaluación del tamaño tumoral y cirugía si una resección anatómica es posible. La resecabilidad puede estar comprometida por multifocalidad, compromiso bilobar, trombosis de vena porta o cava, invasión vascular, linfadenopatias para-aórticas, compromiso del hi lio hepático y metástasis. En general, no se deberían realizar resecciones no anatómicas, pues presentan una alta tasa de recidiva local, en estos casos se debería considerar el trasplante hepático (THO) como opción quirúrgica, actualmente un 6% de los pacientes con hepatoblastoma requerirán para su total resección un THO, la única contraindicación para THO es enfermedad metastásica a distancia. La sobrevida a 10 años, de un hepatoblastoma con resección primaria (con o sin THO) luego de quimioterapia neoadjuvante es de 85%. El THO utilizado como rescate en un paciente luego de recidiva tumoral es de 30 a 50%, por esta razón se sugiere no intentar resecciones tumorales no anatómicas que aumentan el riesgo de finalizar la cirugía con márgenes positivos, y ofrecer a estos pacientes un trasplante hepático como tratamiento quirúrgico primario para la resección del tumor (15). A pesar de que el hepatoblastoma debe ser resecado completamente para su manejo, la quimioterapia sigue siendo fundamental en su tratamiento, existen diferentes protocolos quimioterápicos, el manejo con Cisplatino, Vincristina y 5- Fluoracilo, tienen el mismo pronóstico que el régimen con Cisplatino y Doxorrubicina, el último con menos efectos adversos asociados. La sobrevida del hepatoblastoma ha mejorado en los últimos años, actualmente la sobrevida libre de enfermedad a 5 años es de 100% para estadíos I de histología fetal, 91% estadíos I y II de otras histologías, 64% en estadíos III y 25% en estadíos IV (29).

Sistema De Clasificación Pretext
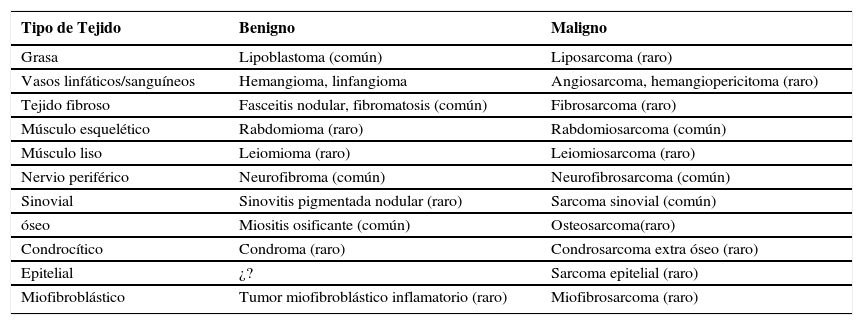
Los tumores que pueden afectar esta área anatómica son múltiples, se dividen en tumores óseo y de partes blandas y dependiendo del origen histológico del cual provengan (Tabla 5). En general los tumores de partes blandas se dividen en rabdomiosarcomas (RMS) y no-rabdomiosarcoma (NRMS), el RMS corresponde a un 50% de todos los tumores de partes blandas. Los tumores óseos malignos más frecuentes son el osteosarcoma y tumor de Ewing. El cuadro clínico, manejo y pronóstico de estos tumores depende del tipo histológico, edad, localización, extensión, resecabilidad y otros factores varios (14).
Tumores De Partes Blandas
| Tipo de Tejido | Benigno | Maligno |
|---|---|---|
| Grasa | Lipoblastoma (común) | Liposarcoma (raro) |
| Vasos linfáticos/sanguíneos | Hemangioma, linfangioma | Angiosarcoma, hemangiopericitoma (raro) |
| Tejido fibroso | Fasceitis nodular, fibromatosis (común) | Fibrosarcoma (raro) |
| Músculo esquelético | Rabdomioma (raro) | Rabdomiosarcoma (común) |
| Músculo liso | Leiomioma (raro) | Leiomiosarcoma (raro) |
| Nervio periférico | Neurofibroma (común) | Neurofibrosarcoma (común) |
| Sinovial | Sinovitis pigmentada nodular (raro) | Sarcoma sinovial (común) |
| óseo | Miositis osificante (común) | Osteosarcoma(raro) |
| Condrocítico | Condroma (raro) | Condrosarcoma extra óseo (raro) |
| Epitelial | ¿? | Sarcoma epitelial (raro) |
| Miofibroblástico | Tumor miofibroblástico inflamatorio (raro) | Miofibrosarcoma (raro) |
Rabdomiosarcomas: El RMS tiene una gran respuesta a quimioterapia, por lo que la cirugía mutilante para lograr su resección total ya no es utilizada. Cuando es posible se debe realizar cirugía primaria del tumor y de los ganglios linfáticos que drenan la lesión si estos están comprometidos, si la cirugía no es posible, se debe tomar una biopsia y comenzar terapia neoadjuvante con resección posterior al tratamiento quimiotera- pico. El manejo depende de la localización del tumor, tipo histológico y posibilidad de resección quirúrgica, esto divide al RMS en tres diferentes grupos de riesgo que determinan el tratamiento sistémico y local: Pacientes de bajo riesgo: 1. RMS embrionarios, localizados en sitios favorables (órbita, cabeza y cuello, no-parameníngeo, genitourinario excepto vejiga y próstata) con enfermedad localizada resecada totalmente reciben Vincristina y Dactinomicina (VA) con o sin RT y 2. RMS embrionarios en sitios no-favorables (vejiga, próstata, extremidades, tronco, abdomen, y sitios parameníngeos) con enfermedad localizada, con o sin enfermedad nodal, totalmente resecado reciben Vincristina, Dactinomicina y Ciclofosfamida (VAC) con o sin RT. Pacientes de riesgo intermedio: RMS alveolar o indiferenciado localizados, independientes del sitio y de la enfermedad nodal y los RMS embrionarios con tumor residual luego de la cirugía o RMS embrionario metastásico en pacientes menores de 10 años de edad, reciben VAC con o sin topotecan y RT. Pacientes de alto riesgo: Pacientes > 10 años con RMS embrionario metastásico, o pacientes < de 21 años con tumores alveolares o indiferenciados metastásico, reciben irinotecan, VAC y RT. Con este manejo multimodal la sobrevida a 5 años es de 71%, 90% para lesiones localizadas, totalmente resecadas (grupo I), 80% para lesiones localizadas resecadas con márgenes positivos microscópicos o lesiones con diseminación nodal totalmente resecadas o tumor residual microscópico en el área de diseminación (grupo II), 70% en tumores resecados en forma incompleta (grupo III) y 30% en pacientes con enfermedad a distancia (grupo IV) (30).
No-Rabdomiosarcomas: Constituyen un grupo diverso de patologías, están definidos por sus características histológicas y anormalidades genéticas que determinan su comportamiento biológico. Los factores pronósticos son tamaño tumoral, resectabilidad, grado histológico, diseminación local y a distancia, a diferencia del RMS, estos tumores tienen mala respuesta a quimioterapia, por lo que el tratamiento quirúrgico, asociado o no a RT constituye el manejo principal para evitar la recurrencia local. El pronóstico para pacientes con tumores diseminados a distancia es 20% a 3 años. En general los tumores localizados, menores de 5 centímetros de diámetro, resecados totalmente con al menos 0,5 centímetro de margen libre, no reciben terapia adyuvante. En resección incompleta, tumores > a 5 centímetros de diámetro y diseminación nodal totalmente resecada, los pacientes reciben RT externa o a través de catéteres de braquiterapia. En tumor no resecable, con enfermedad metastásica, se agrega RT al área comprometida y quimioterapia, diferentes protocolos han sido utilizados con pobre respuesta hasta el momento (31).
Tumores Óseos: La mayoría de los tumores óseos en la infancia son benignos, el osteosarcoma (OSC) corresponde a un 15% de todos los tumores óseos y el tumor de Ewing (TE) a un 4,6%. El OSC tiene su origen en el tejido óseo, pueden ocurrir a cualquier edad, mayormente en adolescentes, puede afectar cualquier hueso pero es más frecuente en las extremidades, siendo las localizaciones más frecuentes el fémur distal, tibia proximal y húmero proximal. Existen diferentes subtipos histológicos que influencian el pronóstico. El TE ocurre a una edad más temprana y también puede afectar cualquier hueso, afectando principalmente el fémur, la pelvis y el húmero. Diferencias clínicas, demográficas y radiológicas nos ayudan a determinar la histología, pero es finalmente la biopsia la que nos entrega la confirmación diagnóstica. El estudio debe evaluar las características y compromiso de estructuras locales y la presencia o no de enfermedad metastásica. El lugar de diseminación más frecuente en OSC son los pulmones y en TE pulmón y médula ósea. Los tumores óseos malignos son tratados con terapia multimodal, los 2 tipos histológicos reciben tratamiento sistémico neoadjuvante, resección tumoral y posterior quimioterapia adyuvante dependiendo de la respuesta a tratamiento en el tejido tumoral y la existencia o no de enfermedad metastásica. La RT ha sido utilizada asociada a quimioterapia con o sin resección quirúrgica en el tratamiento del TE, logrando mejor control local. Cuando es utilizada sin cirugía se deben entregar dosis de 6.600 Gy para lograr control tumoral local, en contraste con 3.000 a 4.000 Gy requeridos cuando es utilizada asociada a resección quirúrgica, factor a considerar cuando se tratan tumores pediátricos de extremidades, en el que el crecimiento del la extremidad puede verse comprometido por el grado de radiación recibida. En tumores óseos la resección y reconstrucción son los elementos principales del manejo quirúrgico, esta terapia debe ser abordada con un equipo multidisciplinario compuesto por cirujanos oncólogos y traumatólogos, fisioterapeutas y radioterapeutas en TE. El objetivo principal es resecar el tumor sin contaminación y con márgenes negativos suficientes y la reconstrucción de tejidos para idealmente preservar la extremidad con buena función motora, esto se logra con la utilización de prótesis y movilización de tejidos adyacentes para cubrir el defecto dejado con la resección, en algunas ocasiones no es posible resecar el tumor preservando la extremidad, en esos pacientes la amputación de la extremidad es requerida para el manejo exitoso del tumor. Ante la presencia de metástasis en OSC, estas se manejan con resección quirúrgica y quimioterapia (32).
ConclusiónLos avances en los métodos diagnósticos de laboratorio y radiológicos, el mayor entendimiento de la genética molecular de los tumores y su comportamiento biológico, asociado al desarrollo de tratamiento farmacológico, radioterapia y de técnicas quirúrgicas han permitido mejorar la sobrevida de los pacientes pediátricos con patología tumoral, sin embargo en la mejoría de los resultados en el cáncer pediátrico es fundamental un diagnóstico precoz, el que se hace difícil debido a que el 85% de los cánceres infantiles se presenta con signos y síntomas inespecíficos. En este artículo entregamos aspectos epidemiológicos, diagnósticos y terapéuticos de la patología quirúrgica oncológica pediátrica, elemento fundamental para realizar una derivación precoz y oportuna.
La autora declara no tener conflictos de interés, en relación a este artículo.