




The “secondary injury” theory of liver failure indicated that hyperammonaemia due to liver failure causes further deterioration of hepatocytes. Our previous studies have demonstrated that high blood ammonia levels may lead to hepatocyte apoptosis, as NH4Cl loading caused metabolic acidosis and an increase in sodium-hydrogen exchanger isoform 1 (NHE1). In this study, we established a hyperammonia hepatocyte model to determine the role of NHE1 in the regulation of hepatocyte apoptosis induced by NH4Cl.
Materials and methodsIn current studies, intracellular pH (pHi) and NHE1 activity were analyzed using the pHi-sensitive dye BCECF-AM. The results showed that intracellular pH dropped and NHE1 activity increased in hepatocytes under NH4Cl treatment. As expected, decreased pHi induced by NH4Cl was associated with increased apoptosis, low cell proliferation and ATP depletion, which was exacerbated by exposure to the NHE1 inhibitor cariporide. We also found that NH4Cl treatment stimulated PI3K and Akt phosphorylation and this effect was considerably reduced by NHE1 inhibition.
ConclusionThis study highlighted the significant role of NHE1 in the regulation of cell apoptosis induced by hyperammonaemia.
La teoría de la «lesión secundaria» de la insuficiencia hepática mostró que la hiperamoniaquemia provocada por la insuficiencia hepática causa mayor deterioro de los hepatocitos. Nuestros anteriores estudios previos han demostrado que los niveles altos de amoníaco en sangre pueden conducir a la apoptosis de los hepatocitos. Como la carga de NH4Cl provocó acidosis metabólica y un aumento de la isoforma 1 del intercambiador de sodio/hidrógeno (NHE1). En este estudio, establecimos un modelo de hepatocitos de hiperamonia para establecer el papel de NHE1 en la regulación de la apoptosis de hepatocitos inducida por NH4Cl.
Materiales y métodosEn los estudios actuales, el pH intracelular (pHi) y la actividad del NHE1 se analizaron con el colorante BCECF-AM, sensible al pHi. Los resultados mostraron que el pH intracelular disminuyó y la actividad del NHE1 aumentó en hepatocitos con tratamiento del NH4Cl. Como se esperaba, la disminución del pHi inducido por NH4Cl se relacionó con un aumento de la apoptosis, baja proliferación celular y reducción del ATP, que se exacerbó por la exposición a cariporide, inhibidor del NHE1. También encontramos que el tratamiento del NH4Cl estimuló la fosforilación de PI3K y Akt, y este efecto se redujo considerablemente por la inhibición del NHE1.
ConclusiónEste trabajo ha destacado el importante papel del NHE1 en la regulación de la apoptosis celular inducida por hiperamoniaquemia.
Hepatic failure (HF) is a life threatening disease and is caused by a variety of factors, which induce liver cell damage and liver dysfunction. The mortality rate of HF is very high1 and the complications associated with HF include hepatic encephalopathy, hepatorenal syndrome and hemorrhaging.2 It has been proved that hyperammonaemia was involved in the pathogenesis of hepatic encephalopathy and it is commonly studied as the mechanism of encephalopathy.3
Recent studies, including the results obtained from our laboratory, have demonstrated that hyperammonemia has a direct adverse effect on hepatocytes and it is therefore both a cause and an effect of hepatic failure.4–6 But the effect of high blood ammonia on liver cell injury and its underlying mechanism remain unclear.
Proper cell function depends on the maintenance of intracellular and extracellular physicochemical parameters within certain physiological limits. Such an important parameter is pH.7 In fact, the activity of intracellular enzymes, the interaction of cytoskeletal elements, the rate at which cells grow and differentiate all depend on pHi.8,9 Cells can minimize significant pHi fluctuations through several H+ transport systems; the best known system is represented by the sodium/hydrogen exchanger (NHE) family. Among all the NHE isoforms, NHE isoform 1 (NHE1) is the dominant subtype in hepatic tissue and displays a widespread tissue distribution by maintaining pHi and cell volume levels, in contrast, other subtypes have more limited tissue distribution and are thought to be involved in NaCl absorption.11,12
Most studies have demonstrated that apoptosis is ultimately accompanied by cytosol acidification.10,13,14 More recently, it was reported that rats subjected to 280mmol/L NH4Cl loading for five days developed metabolic acidosis.15 Flow cytometric analysis from our previous research work revealed that the cell apoptosis ratio increased with increasing concentrations of NH4Cl. This demonstrated that high blood ammonia levels may lead to liver cell damage and apoptosis.4–6,16 A cytoprotective role of NHE1 is also supported by experiments in which NHE1-null proximal tubule cells demonstrated enhanced sensitivity to multiple apoptotic stimuli compared with wild-type cells.14 Manucha17 demonstrated that tubular epithelial cell apoptosis was associated with decreased NHE1 expression in a neonatal rat model of ureteral obstruction.
In our present study, we investigated whether hyperammonemia has any effects on the pHi and activity of NHE1 in hepatic cells. A hyperammonia hepatic cell model was established to determined the role of NHE1 in the regulation of hepatocyte apoptosis induced by hyperammonaemia and its mechanism underlying liver failure.
Materials and methodsMaterialsAll cell culture reagents, including antibiotics, fetal bovine serum (FBS), phosphate-buffered saline (PBS) and RIMP1640 were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). 2′,7′-bis(2-carboxyethyl)-5(6)-carboxyfluo rescein acetoxymethyl ester (BCECF-AM), cariporide and antibodies were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Cell culture plastics were purchased from Corning Incorporated (Corning, New York, USA). Annexin V fluorescein isothiocyanate (FITC) apoptosis detection kit was purchased from Beyotime Institute of Biotechnology (Haimen, China). All other chemicals were purchased from Sigma–Aldrich (St. Louis, MO, USA).
Cell line and experimental treatmentsHepatocytes were obtained from the Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. When the cells monolayer became confluent, the cells were divided into four groups and incubated for 12h in serum free medium or serum free medium with 10μM cariporide, 5mM NH4Cl, or 5mM NH4Cl+10μM cariporide, respectively.
Intracellular pHi and NHE activity measurementHepatocytes were plated on glass coverslips and in complete RIMP1640 at 37°C in a humidified atmosphere with 5% CO2. Intracellular pH was measured after chemical treatments. Cellular NHE activity was determined fluorometrically by using intracellular pH-sensitive dye acetoxymethyl ester of BCECF-AM, as described previously.10 In briefly, cells were exposed to Na+ solution (138mM NaCl, 5mM KCl, 2mM CaCl2, 1mM NaH2PO4, 1mM MgSO4, 25mM glucose, and 20mM HEPES, pH 7.4) with 5μM BCECF-AM at 37°C for 1h. Then cells were washed with Na+ solution to remove extracellular dye and perfused with Na+ solution and measured the baseline pHi.
Next, cells were exposed to 40mM NH4Cl (pH 7.4) and perfused with Na+ free medium (130mM tetramethyl ammonium chloride, 5mM KCl, 2mM CaCl2, 1mM NaH2PO4, 1mM MgSO4, 25mM glucose, and 20mM HEPES, pH 7.4). Then, re-addition of extracellular Na+ allowed activation of Na+/H+ exchange and recovery from acidification to basal levels. At the end of each experiment, the fluorescence ratio was calibrated to pHi by using nigericin (2μM)/high K+ solutions of different pH values method.
Initial rate of Na+-dependent recovery from intracellular acidification corresponding to NHE1 activity was almost linear during the first 1min of the reaction. NHE1 activity was measured by calculating the pHi change over the first 1min and expressed as ΔpH/min.
Cell viability assayAfter experimental treatments, cell viability of hepatocytes was demonstrated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay according to manufacturer's instructions. In brief, MTT solution (5mg/ml) was added to each well and incubated for 4h at 37°C. Then 1ml of dimethyl sulfoxide (DMSO) was added and thoroughly mixed for 10min. Subsequently, MTT absorbance was measured at 570 and 630nm using a microplate reader.
Flow cytometryTo confirm the results from MTT assay, hepatocytes were harvested using trypsin/EDTA solution after experimental treatments, rinsed with binding buffer (0.01M HEPES, 2.5mM CaCl2, 0.14M NaCl, pH 7.4) and collected by centrifugation at 1000×g for 10min. Cell pellets were resuspended with binding buffer at a concentration of 1×106cells/ml and incubated with propidium iodide and FITC-Annexin V following the manufacturer's recommendations for 15min at room temperature in the dark. Cell apoptosis analysis was performed by using a FACScalibur (Becton Dickinson Corp., USA).
Measuring intracellular ATP contentBriefly, following cell preparations, intracellular ATP content was measured as described previously18 by bioluminescence assay using a commercial ATP assay kit (EMD Millipore, Darmstadt, Germany) according to the manufacturer's instructions. In brief, cells were treated with 100μL nuclear releasing reagent for 5min at room temperature while gently shaking, and 1.0μL ATP monitoring enzyme was added into the cell lysates. Bioluminescence of intracellular ATP content was determined by comparing with a standard curve that was generated by using various ATP solutions with known concentrations. The intracellular ATP is further normalized to protein concentration for comparison.
Western blot analysisFollowing experimental treatments, hepatocytes were washed three times by ice-cold PBS containing 50mM Tris. Then hepatocytes were harvested by centrifugation at 750×g for 10min at 4°C. Cell pellets were resuspended and lysed in 500μl of ice-cold lysis buffer (50mM NaCl, 10mM HEPES, 5mM EDTA, 1mM benzamidine, 0.5% Triton X-100). Cell lysate was solubilized at 4°C for 30min with end-over-end rotation and subsequently homogenized 10 times using a 26-gauge needle applied to a 1-ml syringe. Cellular debris was cleared by centrifugation at 14,000×g for 15min. Supernatant was collected and solubilized in loading buffer (5mM Tris–HCl, 1% SDS, 10% glycerol, and 1% 2-mercaptoethanol, pH 6.8), boiled for 10min. Protein content of each fraction was determined by the bicinchoninic acid assay (Sigma). Subsequently, normalized samples were loaded, size-fractionated by 10% SDS-PAGE, and electrophoretically transferred to nitrocellulose membrane. After blocking with 5% nonfat milk in PBST (0.05% Tween/PBS) at room temperature for 1h and incubated with anti-p-Akt antibody, anti-Akt antibody, anti-p-PI3K antibody and anti-PI3K antibody or anti-β-actin antibody (dilution 1:1000), respectively, membrane was washed with PBST for three times for 10min each. Following incubation with Alexa Fluor®488 mouse anti-rabbit IgG and Alexa Fluor® 680 rabbits anti-mouse IgG (Thermo Fisher Scientific, Inc.; dilution 1:10,000) for 60min at room temperature in the dark. Subsequently, lots were detected and relative density units were estimated from the mean pixel density using Image Studio Lite 4.0 and normalized to β-actin.
Statistical analysisStatistical analyses were performed using SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA). Data from multiple experiments were processed and expressed as mean±standard deviation. Statistical differences between the different groups were determined using one-way analysis of variance followed by a Student–Newman–Keuls test. P<0.05 was considered to indicate a statistically significant difference.
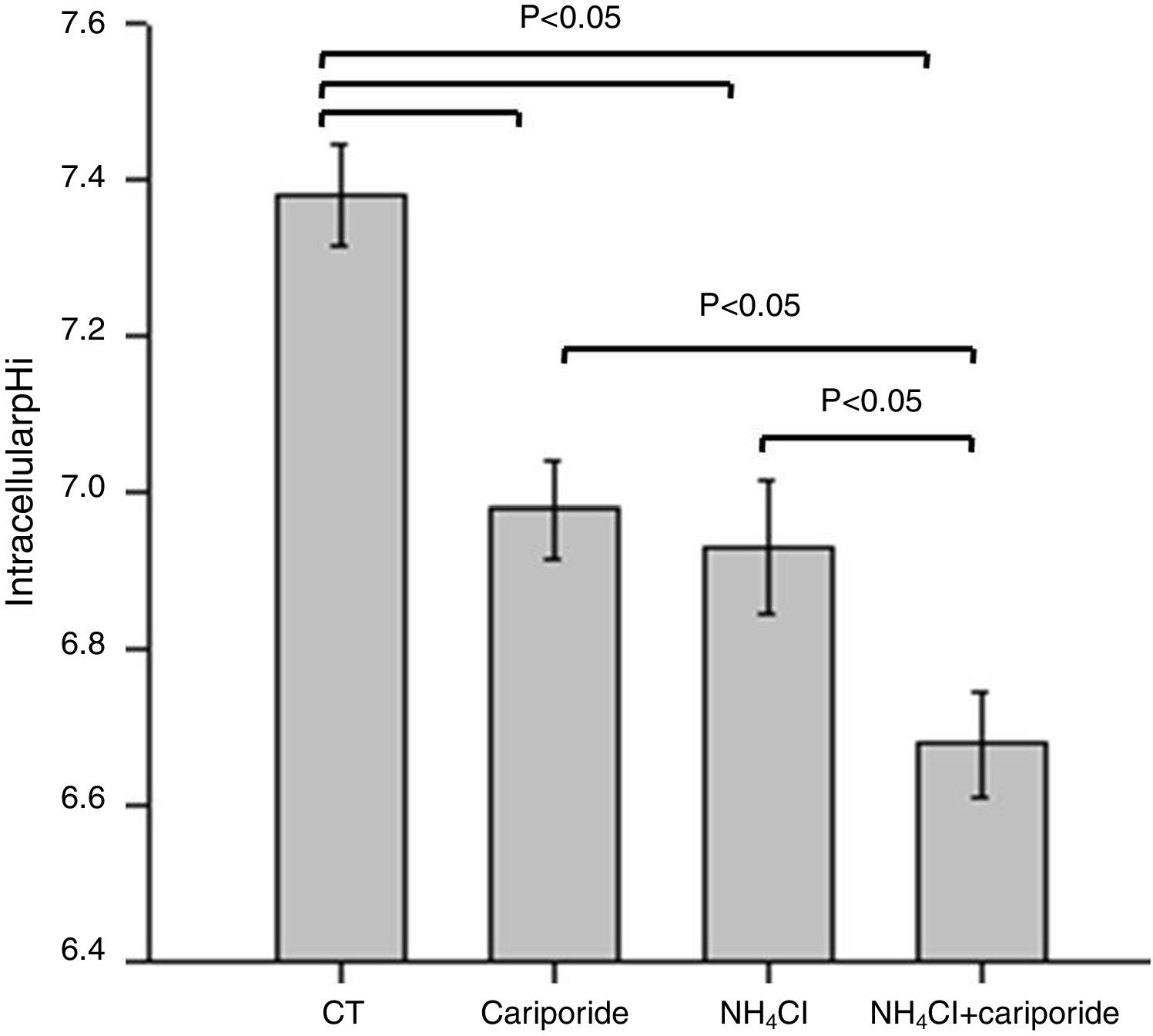
ResultsNH4Cl and cariporide lowered intracellular pHTo verify the effects of NH4Cl and cariporide on intracellular pH in hepatocytes, the fluorescent indicator dye BCECF-AM was used. Fig. 1 showed that application of NH4Cl produced an obvious intracellular acidification. As expected, cariporide, a specific NHE1 inhibitor, decreased pHi, but its effect on pHi was much less than that in NH4Cl medium. However, this intracellular acidification was synergistically increased when they were used together.
. Hepatic cells were treated for 24h with NH4Cl, cariporide or NH4Cl plus cariporide. Control cells were untreated. The intracellular pH (pHi) was then determined with the use of BCECF-AM as described under “Materials and methods”. Results are presented as the mean±standard deviation of three independent experiments and samples were analyzed in triplicate.")
Intracellular acidification upon treatment with NH4Cl and cariporide (x¯±sd, n=9). Hepatic cells were treated for 24h with NH4Cl, cariporide or NH4Cl plus cariporide. Control cells were untreated. The intracellular pH (pHi) was then determined with the use of BCECF-AM as described under “Materials and methods”. Results are presented as the mean±standard deviation of three independent experiments and samples were analyzed in triplicate.
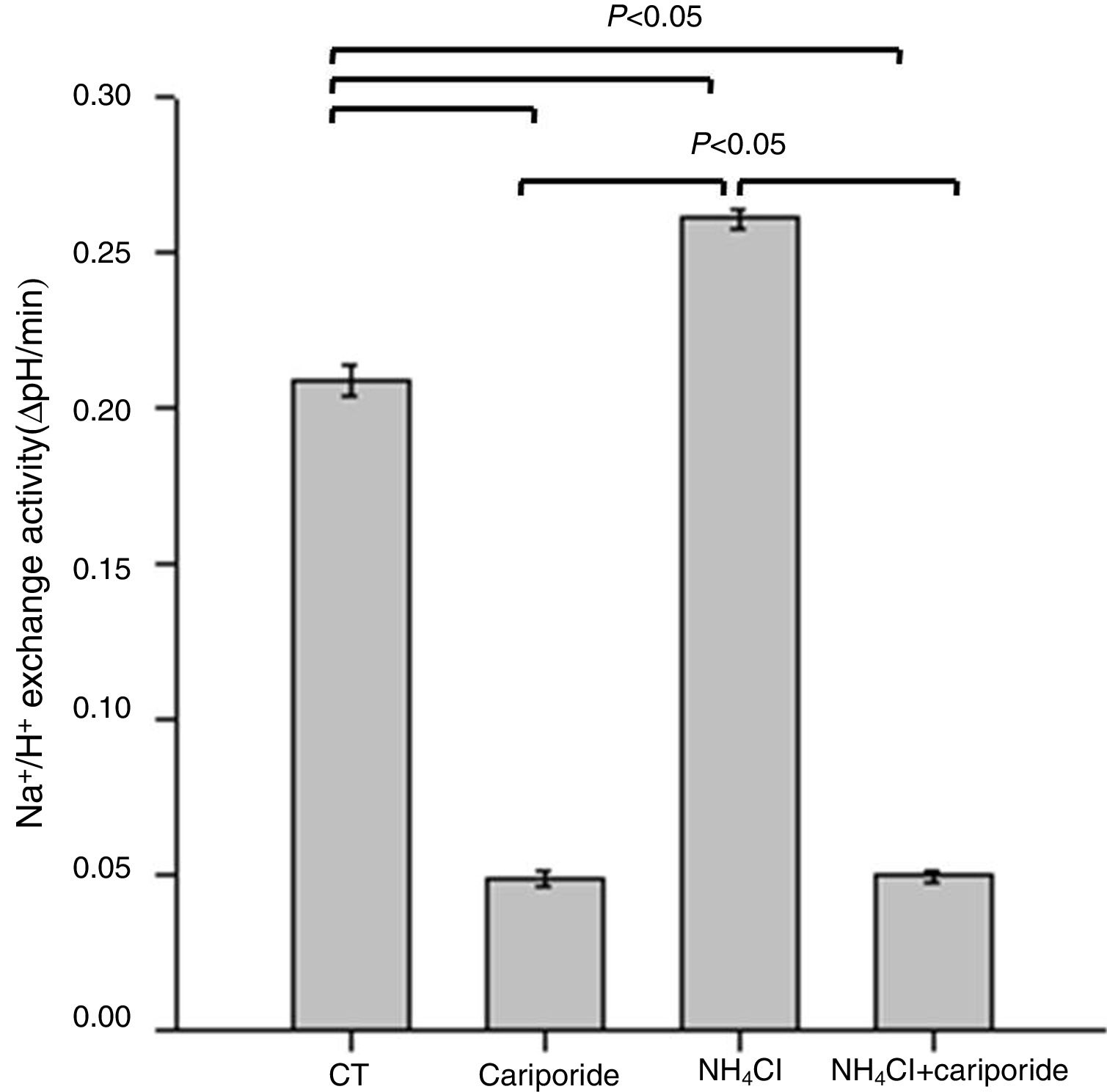
Because NHE activity contributes to the maintenance of intracellular pH homeostasis, we next investigated the effects of NH4Cl and cariporide on NHE1 activity. In hepatic cells, the mean activity of the NHE1 was 0.209±0.005 ΔpH/min and more than 77% of the sodium dependent pHi recovery is inhibited by cariporide. When hepatic cells were incubated with 5mM NH4Cl, Na+/H+ activity significantly increased to 0.261±0.003 ΔpH/min, whereas addition of cariporide to the culture medium with 5mM NH4Cl decreased Na+/H+ activity to the same level as the cariporide group (Fig. 2).
. Hepatic cells were treated for 24h with NH4Cl, cariporide or NH4Cl plus cariporide, respectively. Control cells were untreated. NHE activity was measured by calculating the pHi change over the first 1min and expressed as ΔpH/min with the use of BCECF-AM as described under “Materials and methods”. Results are presented as the mean±standard deviation of three independent experiments and samples were analyzed in triplicate.")
Effects of NH4Cl and cariporide on NHE1 Activity (x¯±sd, n=9). Hepatic cells were treated for 24h with NH4Cl, cariporide or NH4Cl plus cariporide, respectively. Control cells were untreated. NHE activity was measured by calculating the pHi change over the first 1min and expressed as ΔpH/min with the use of BCECF-AM as described under “Materials and methods”. Results are presented as the mean±standard deviation of three independent experiments and samples were analyzed in triplicate.
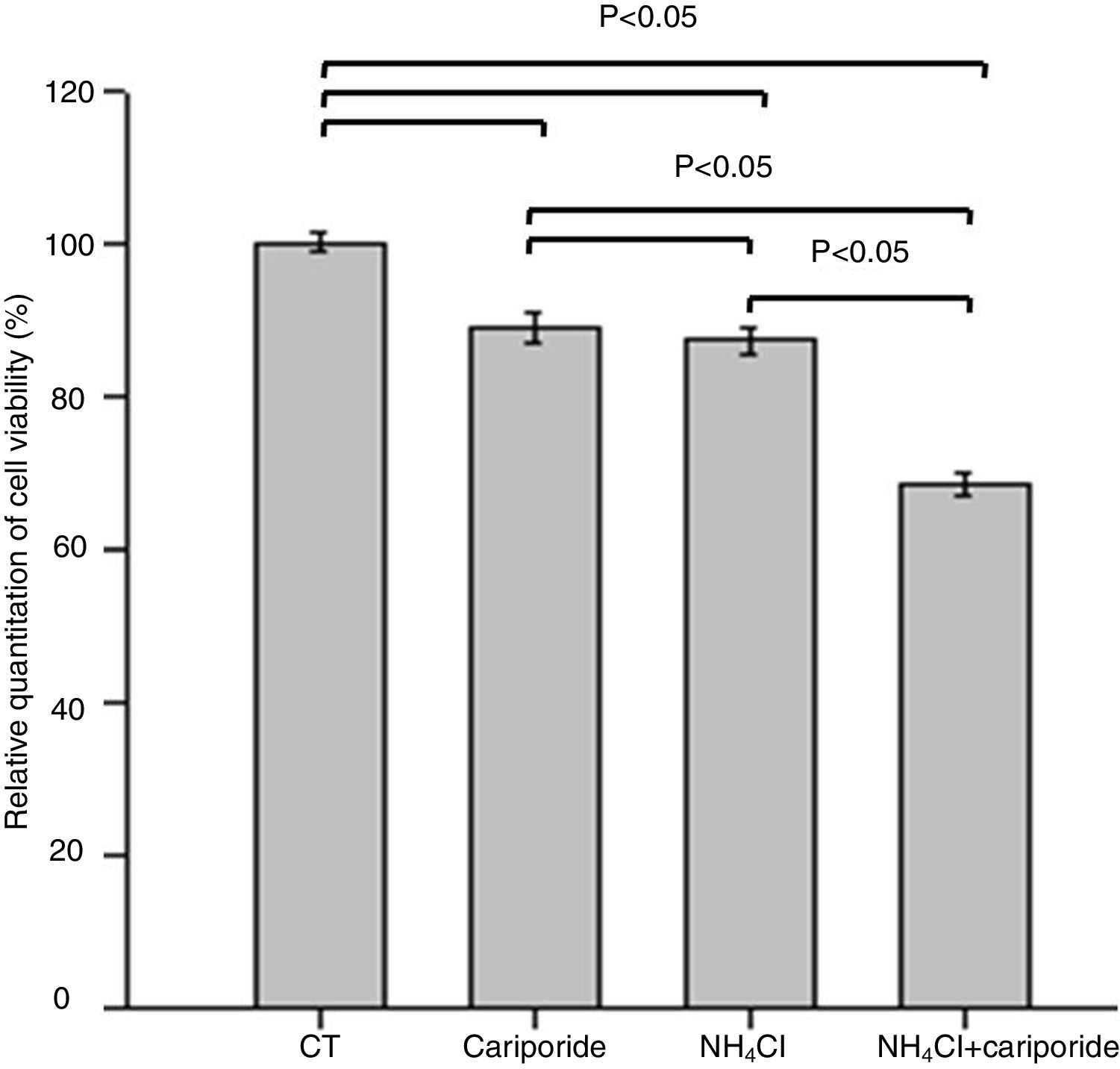
The effects of NH4Cl on the viability of hepatic cells were valuated by MTT assay. The data revealed that treatment of hepatic cells with cariporide or 5mM NH4Cl reduced the cell viability by 10.88% or 12.69% of the control cells, respectively. Whereas cell viability decreased significantly to 68.44% when they were used together (Fig. 3).
. Hepatic cells were treated for 24h with NH4Cl, cariporide or NH4Cl plus cariporide, respectively. Control cells were untreated. Cell viability was determined by MTT assay as described under “Materials and methods”, and was presented as optical density versus control. Results are presented as the mean±standard deviation of three independent experiments and samples were analyzed in triplicate.")
Cell viability upon treatment with NH4Cl and cariporide (x¯±sd, n=9). Hepatic cells were treated for 24h with NH4Cl, cariporide or NH4Cl plus cariporide, respectively. Control cells were untreated. Cell viability was determined by MTT assay as described under “Materials and methods”, and was presented as optical density versus control. Results are presented as the mean±standard deviation of three independent experiments and samples were analyzed in triplicate.
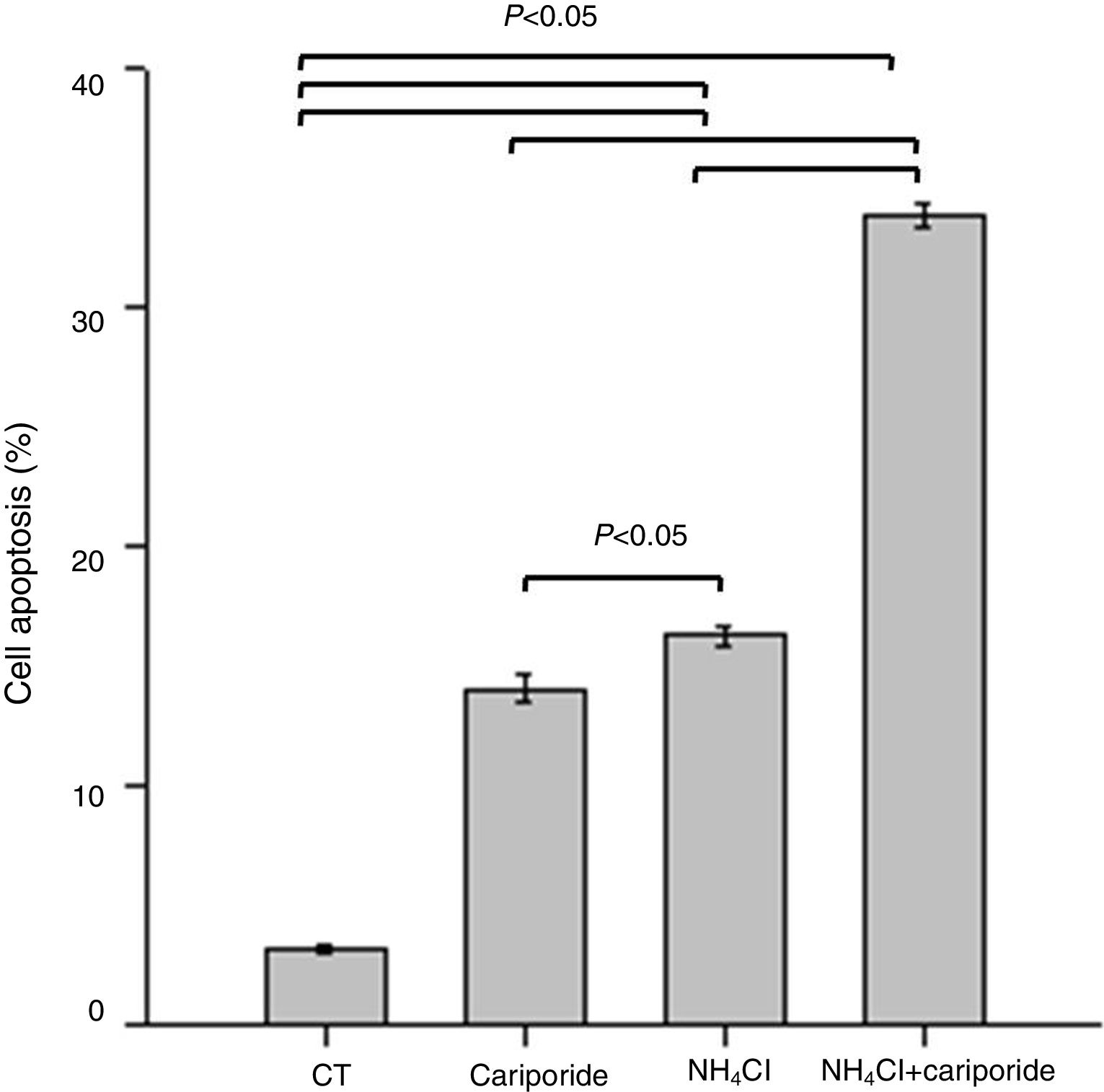
Because above data suggests good correlation between decreased pHi and increased hepatocytes death, the involvement of apoptosis in the hepatocytes death induced by NH4Cl is evaluated by flow cytometry analysis. After 24h treatment with NH4Cl or cariporide increased cell apoptosis up to 14.03±0.56% or 16.27±0.38%, respectively, and interestingly, co-incubation with cariporide increased cell apoptosis dramatically in ammonia-treated cells to almost 33.88±0.53% compared with the control (3.16±0.17%) (Fig. 4).
. Hepatic cells were treated for 24h with NH4Cl, cariporide or NH4Cl plus cariporide, respectively. Control cells were untreated. Cell injury was determined as described under “Materials and methods”, and was presented as the percentage of cells with FITC-Annexin V binding excluding propidium iodide staining in total cell numbers. Results are presented as the mean±standard deviation of three independent experiments and samples were analyzed in triplicate.")
Cell apoptosis upon treatment with NH4Cl and cariporide (x¯±sd, n=9). Hepatic cells were treated for 24h with NH4Cl, cariporide or NH4Cl plus cariporide, respectively. Control cells were untreated. Cell injury was determined as described under “Materials and methods”, and was presented as the percentage of cells with FITC-Annexin V binding excluding propidium iodide staining in total cell numbers. Results are presented as the mean±standard deviation of three independent experiments and samples were analyzed in triplicate.
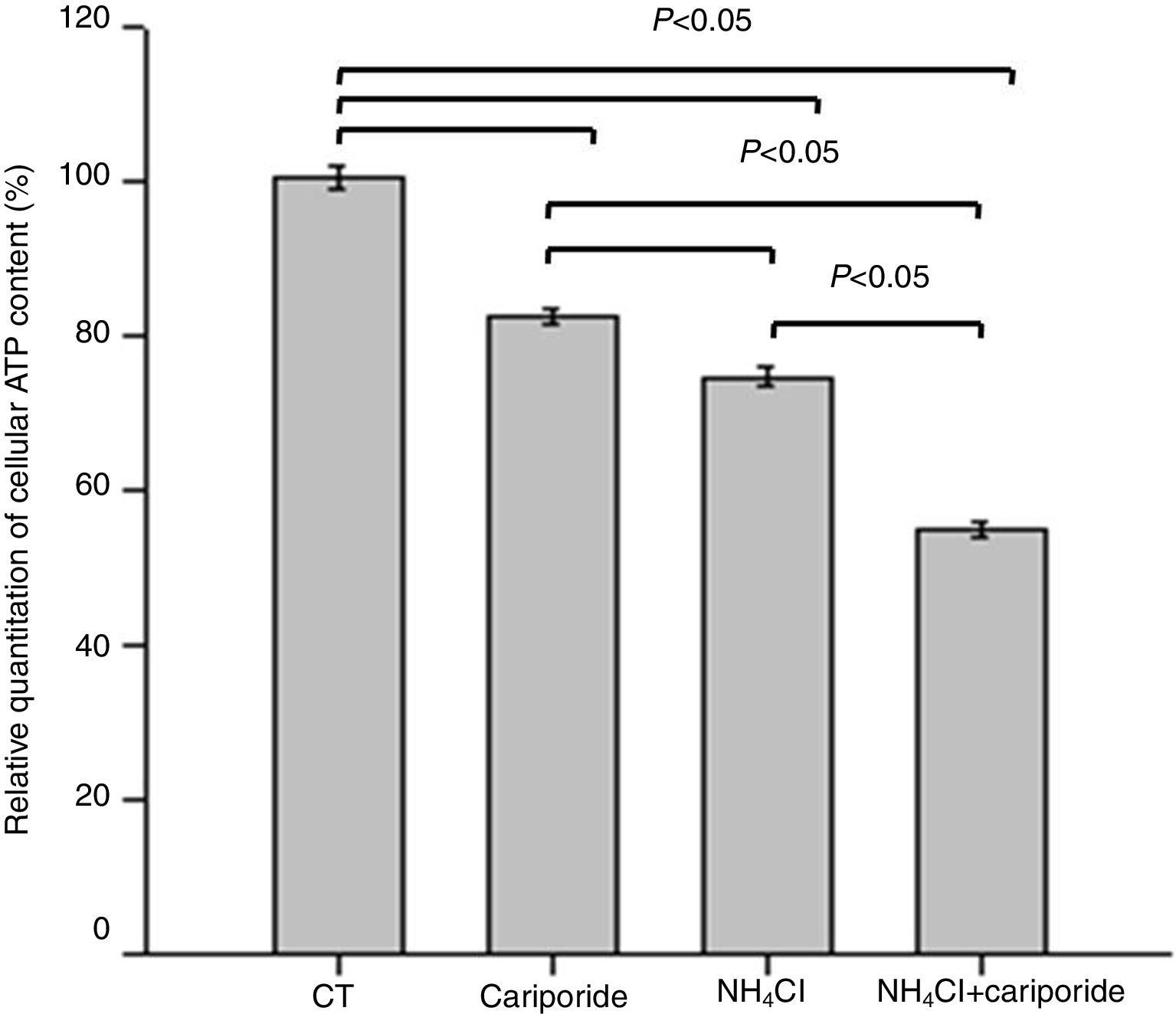
As apoptosis is an ATP-dependent process, we next investigated the effects of NH4Cl and cariporide on cellular ATP. After 24h of treatment with cariporide, cellular ATP in hepatic cells was decreased to 82%, whereas treatment of hepatic cells with 5mM NH4Cl reduced cellular ATP to 74.59±1.28%, and addition of cariporide to the culture medium with 5mM NH4Cl decreased cellular ATP even more to 55% compared with the control (Fig. 5).
. Hepatic cells were treated for 24h with NH4Cl, cariporide or NH4Cl plus cariporide, respectively. Control cells were untreated. Cellular ATP was determined as described under “Materials and methods”. Values are presented as the percentage vs. control. Results are presented as the mean±standard deviation of three independent experiments and samples were analyzed in triplicate.")
Effects of NH4Cl and cariporide on cellular ATP (x¯±sd, n=9). Hepatic cells were treated for 24h with NH4Cl, cariporide or NH4Cl plus cariporide, respectively. Control cells were untreated. Cellular ATP was determined as described under “Materials and methods”. Values are presented as the percentage vs. control. Results are presented as the mean±standard deviation of three independent experiments and samples were analyzed in triplicate.
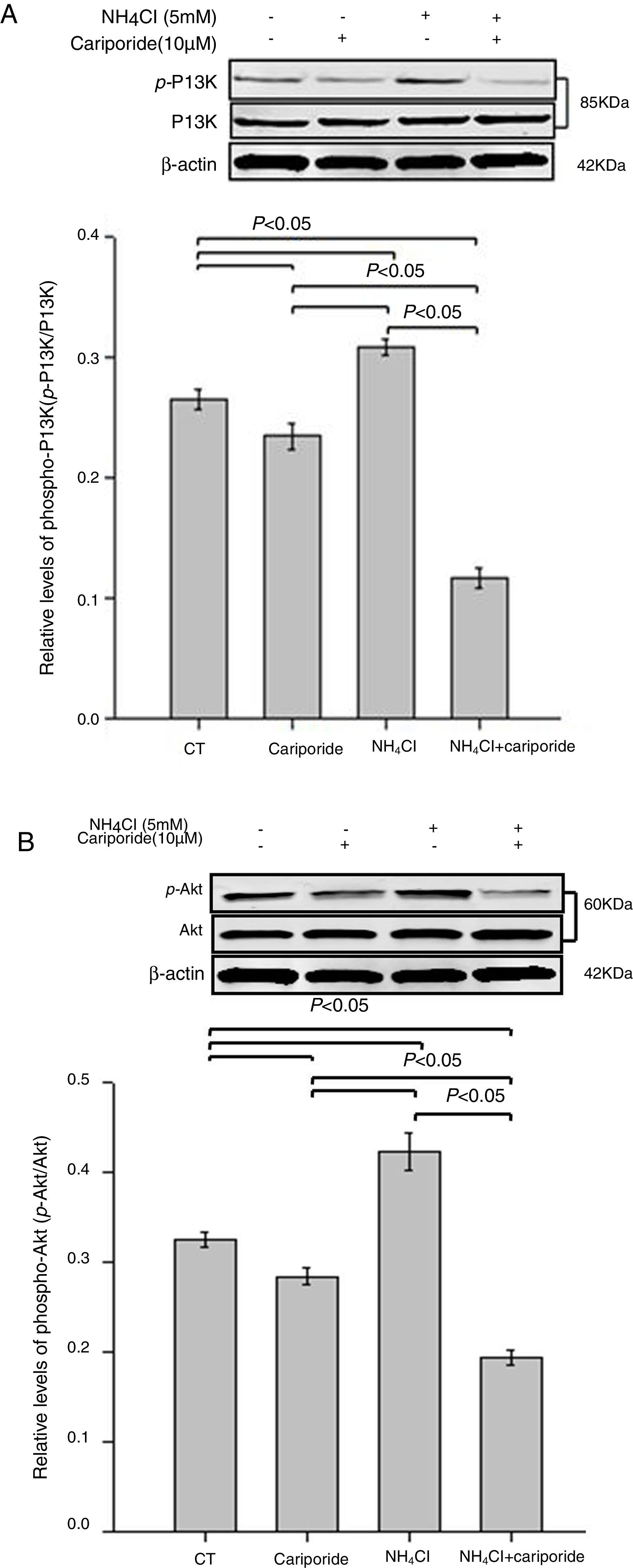
As the PI3K/Akt pathway is a well-known antiapoptotic pathway, we examined whether NH4Cl and cariporide affected the kinase-mediated phosphorylation process in the following experiments. The relative levels of p-Akt/Akt and p-PI3K/PI3K were monitored by western blot analysis. Fig. 6 shows that cariporide slightly reduced the ratio of p-Akt/Akt and p-PI3K/PI3K, whereas, the ratio of p-Akt/Akt and p-PI3K/PI3K significantly increased in NH4Cl treated cells and the upregulated phosphorylation of Akt and PI3K was strongly blocked by cariporide.
. Hepatic cells were treated for 24h with NH4Cl, cariporide or NH4Cl plus cariporide, respectively. Control cells were untreated. Normalized samples were determined by western blot analysis using β-actin as the loading control. Values were presented as ratios of p-Akt/Akt or p-PI3K/PI3K. Results are presented as the mean±standard deviation of three independent experiments.")
Effects of NH4Cl and cariporide on p-Akt/Akt and p-PI3K/PI3K (x¯±sd, n=3). Hepatic cells were treated for 24h with NH4Cl, cariporide or NH4Cl plus cariporide, respectively. Control cells were untreated. Normalized samples were determined by western blot analysis using β-actin as the loading control. Values were presented as ratios of p-Akt/Akt or p-PI3K/PI3K. Results are presented as the mean±standard deviation of three independent experiments.
The ‘secondary injury’ theory of HF indicates that serious metabolic disorders develop, including hyperammonaemia, lactic acid poisoning, blood disorders and increased indole levels, and hyperammonaemia is the most significant causes of liver abnormalities or failure.18,19 Many studies supported that hyperammonaemia, which is both the result and the cause of liver damage, may aggravate hepatic damage.4–6,16
Previous studies from ours showed that hyperammonemia induce liver cell damage, not by inflammation or necrosis, but by cell apoptosis. Cell apoptosis was the predominant effect after treating with low concentration of NH4Cl for a short period of time. As the concentration of NH4Cl increased and the treatment time lengthened, the number of apoptotic cells increased significantly and the growth of cells was markedly inhibited.4,16
It has been reported that rats subjected to NH4Cl loading developed metabolic acidosis15 and metabolic acidosis increased NHE1 activity/expression.20 Intracellular pH is a fundamental modulator of protein function in cells. Optimum pH for endonuclease and caspase activation are 6.3–6.8 and intracellular acidification is critical for apoptosis execution.21 Activation of the NHE1 results in an efflux of H+ and influx of Na+ ions with a resultant increase in pHi, and NHE1-induced cytosol alkalinization were sufficient to prevent apoptosis. Additionally, cells undergoing apoptosis exhibit persistent cell shrinkage, even under isotonic environments. NHE1-mediated cytoplasmic volume expansion by inducing Na+ and H2O influx also protect cells against apoptotic volume decreases.22
Cariporide, which is a highly selective NHE-1 inhibitor with no apparent effect on the Na+/Ca2+ exchanger or fast Na+ currents at levels not exceeding 10μmol/l,23 NHE1 inhibition induced by cariporide has been implicated as an explanation for diminished intracellular pH (pHi), with possible mechanisms of inactivation including ATP depletion, NHE1 dephosphorylation, or cleavage.14 Cariporide was used in this study because it specifically inhibits the NHE-1 exchanger.
It has been well known that pHi may contribute to NHE1-mediated modulation of cell proliferation and cells cannot properly differentiate if lacking NHE1.24,25 Suppression of apoptosis can be achieved by NHE1 activation and cellular alkalinization, that is stimulated by phosphorylation of the C-terminal regulatory domain of the NHE resulting in increased affinity for intracellular H+.26 In this study, we noted NH4Cl treatment caused cytoplasmic acidification, and in response, the activity of NHE1increased. On the contrary, cariporide inhibited the activities of NHE1 and reduced pHi in hepatic cells. And we also noted that the presence of cariporide decreased the activity of NHE-1induced by NH4Cl and enhanced cytoplasmic acidification. As expected, we also observed that decreased pHi induced by NH4Cl was associated with increased apoptosis and low cell proliferation, which was exacerbated by exposure to the NHE1 inhibitor cariporide.
It was reported that NHE inhibitors acted as ATP analogs that caused the formation of nonproductive enzyme–substrate complexes.27 As apoptosis is an ATP-dependent process, we also found the degree of ATP depletion increased when the cells were treated by NH4Cl or cariporide, and when they worked together, the degree of ATP depletion became further lower.
All of these findings are consistent with observations that intracellular acidification triggers cell death.20 NHE1 is likely to be activated in response to decreased pHi and activited NHE1 may then be sufficient to prevent further intracellular acidification and perhaps even promote cell survival, provide the apoptotic stimulus is not too robust.21 Thus it appears that endogenous NHE activity contributes to the maintenance of intracellular pH homeostasis and NHE1 plays a cytoprotective role in mechanics of cell apoptosis. On the contrary, pharmacological inhibition of NHE1 is sufficient to induce or exacerbate apoptotic cell death.24
The PI3K/Akt pathway is a well-known antiapoptotic pathway. Phosphorylated Akt, active form of Akt, activates antiapoptotic proteins such as Bcl-2 and Bcl-xL28 and prevents cytochrome c leakage from the mitochondria.22,29 The present study showed that there is an decrease in phospho-PI3K and phospho-Akt in cariporide medium compared with that in normal medium. On the contrary, ratios of p-Akt/Akt or p-PI3K/PI3K significantly increased under NH4Cl treatment and their stimulation were strongly prevented by cariporide treatment. In this context, upregulation of PI3K/Akt pathway activity appears to play a major role in facilitating cell growth and protecting cells from NH4Cl-induced apoptosis, but dephosphorylation (inactivation) is responsible for the cariporide -induced enhancement of NH4Cl cytotoxicity.
To further clarify the role of the PI3K/Akt pathway linking NHE1 activity, Newell KJ examined NHE1 activity in the presence of three different concentrations of the Akt inhibitor API-2 and found that these inhibitors concentrations had no effect on the rate of acid efflux, thus indicating that Akt is clearly downstream of NHE1. Moreover they also found the decrease in NHE1 activities after cariporide treatment seemed to contribute to reducing ERK phosphorylation.30,31 These findings are consistent with that NHE1 uses ERM to bind PIP2 in a signalplex, which induces PI3K expression. PI3K converts PIP2 into phosphatidylinositol (3,4,5)-phosphate [PI(3,4,5) P3] and results in the phosphorylation of Akt.22 Moreover, the mechanism of blocking the kinase-mediated phosphorylaton process was by competing with ATP.24
An important and novel observation in this study was that intracellular acidification induced by NH4Cl in hepatic cells, stimulated PI3K and Akt phosphorylation and that this effect was strongly reduced by NHE1 inhibitions. This work has highlighted an important role for NHE1 in the protection of hepatocytes from apoptosis induced by hyperammonaemia. Cariporide is an inhibitor of NHE1 and increases apoptosis it is not clinical beneficious, at least in this condition. So, in medical point of view, it will be interesting to suggest strategies to preserve or increase NHE1 activity that may be therapeutically beneficial for acute liver failure.
Taken together, in light of these findings, we postulate that apoptotic stress triggers NHE1-dependent defense against intracellular acidification, but we should realize that NH4Cl-induced protons leaving the hepatic cells via NHE1 may lead to Na+loading, which may subsequently induce Ca2+ overloading as Na+ leaves the cell via the Na+/Ca2+ exchanger if ammonium is cleared out in time. The resultant rise in intracellular calcium concentration is believed to trigger Ca2+-activated protease and phospholipases that cause cellular damage.32 Those are just speculation, which need further research to confirm. Therefore, NHE1-dependent defense against intracellular acidification plays a role in the protective effect in the early stages of apoptosis induced by hyperammonaemia.
ConclusionIncreased levels of ammonia in the blood may be an important cause of further damage to residual liver cells following liver failure. Intracellular acidification induced by NH4Cl triggers cell death and cell apoptosis was the predominant effect in the early injury of liver cells, induced by high levels of blood ammonia. The injury of hepatocytes induced by high ammonia may be related to the reduction of pHi, and increased activation of NHE1 protein in hepatocytes via an increase in phosphorylated Akt to suppress hepatocytes apoptosis, while cariporide inhibits NHE1 activities and amplifies the intracellular acidosis and consequently enhance NH4Cl-induced cytotoxicity via diminution of Akt phosphorylation, verified that suppression of apoptosis can be achieved by NHE1 activation and cellular alkalinization. Overall, our model may provide important insights into how NHE1 plays a role in the protective effect in the early stages of apoptosis induced by hyperammonaemia and we believe that this model provides a framework for future studies.
Conflicts of interestThe authors declare that they have no conflicts of interest.
This work was supported by grants from the National Natural Science Foundation of China (grant no. 81500433), the Foundation of the Henan Educational Committee (grant no. 16A310003), the Science and Technology Planning Project of Henan (grant nos. 132102310138 and 142102310140), the Medical Science and Technology Projects of Henan, Doctoral Entrepreneurs Foundation and Health Science and Technology Innovation Talents Foundation of Henan.
The authors are grateful for the financial support to be able to perform this work.

