




La sarcoidosis es una enfermedad crónica multisistémica de etiología desconocida, caracterizada por el acúmulo de células inflamatorias con formación de granulomas epiteloides no caseificantes debido a una respuesta inflamatoria celular excesiva1,2. El pulmón y los ganglios linfáticos de los hilios pulmonares son los órganos más frecuentemente afectados (90% de casos), aunque puede verse involucrado cualquier órgano2. Es más frecuente en mujeres jóvenes de raza negra y tiene una prevalencia de 10-40 casos por 100.000 personas. La afectación hepática aislada es rara, y la forma de presentación clínica, variable1–4. Para el diagnóstico de certeza es necesario, junto con los datos clínicos, de laboratorio y radiológicos compatibles, una biopsia hepática que muestre granulomas no caseificantes típicos5. Presentamos un caso de colestasis intrahepática recurrente secundaria a sarcoidosis hepática (SH) aislada confirmada histológicamente mediante biopsia, con buena respuesta al tratamiento esteroideo. Se realiza, además, una revisión de las manifestaciones clínicas, el diagnóstico y el tratamiento de la SH.
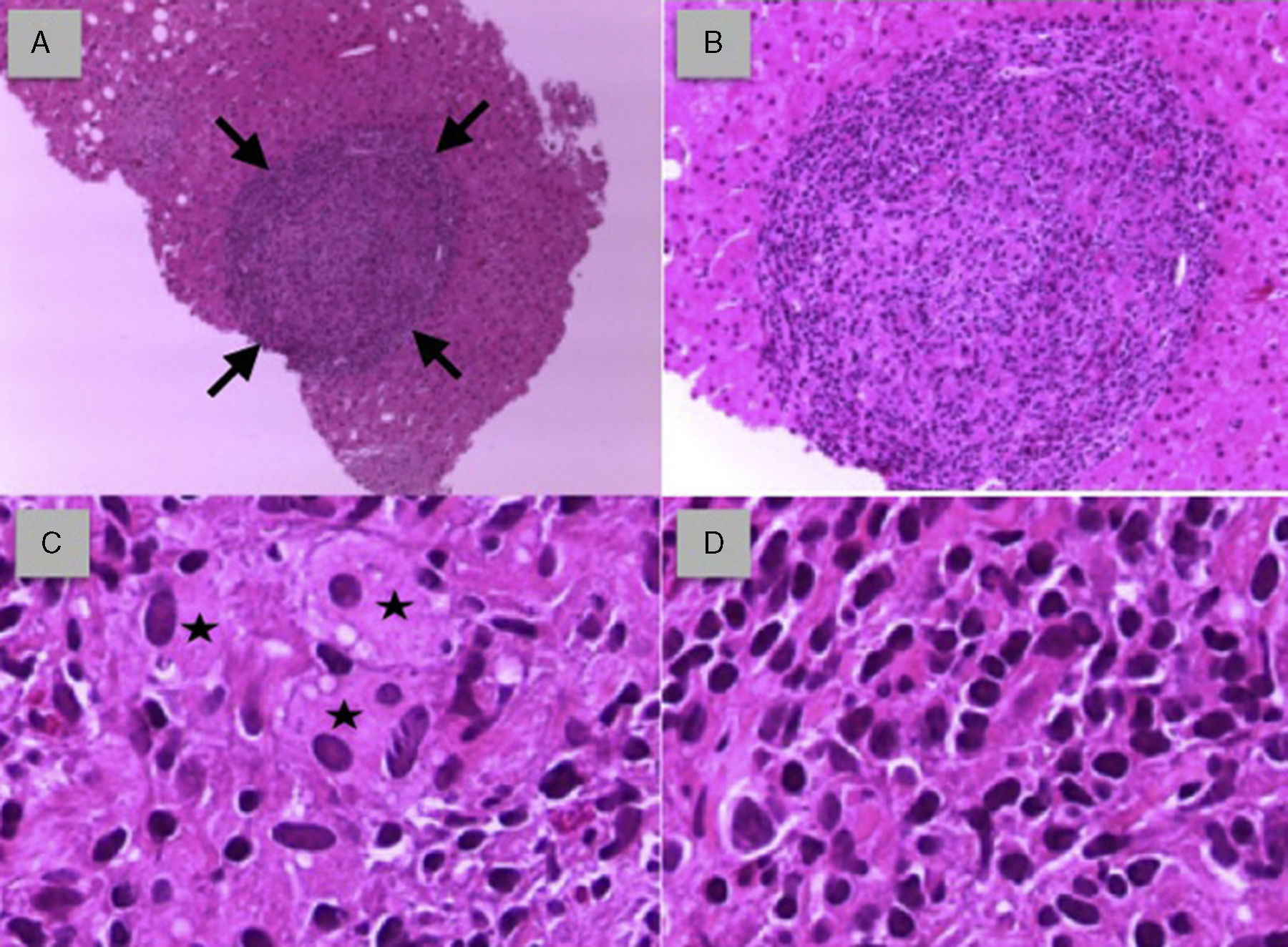
Presentamos el caso de una mujer de 72 años, fumadora de 30 paquetes/año y EPOC con obstrucción severa al flujo aéreo en tratamiento con broncodilatadores inhalados. Acude a la consulta de atención primaria por síndrome constitucional de 2 meses de evolución, junto con fiebre y molestias abdominales inespecíficas. Un año antes ingresó en el hospital por un episodio de similares características, autolimitado, sin diagnóstico tras estudio amplio. En la exploración física se encontraba febril (38,2°C), con taquipnea superficial, discreta palidez cutánea, sin adenopatías, y a la palpación abdominal presentaba hepatomegalia dolorosa de 2 traveses de dedo. Se solicitó analítica inicial, que mostraba discreta anemia (Hb 10,2mg/dl), linfopenia (980cél/mm3), hipercalcemia leve (10,8mg/dl), aumento de reactantes de fase aguda (VSG 56mm/h, PCR 216mg/dl, ferritina 1.216ug/l), enzima convertidora de angiotensina elevada (99ug/l) y patrón de colestasis (FA 246UI/l, GGT 547U/I, con AST y ALT normales). Los marcadores tumorales, el estudio de autoinmunidad (ANA, ENA, AMA, AML, anti-LKM, anti-LC-1, SLA/LP), la serología (incluidos virus hepatotropos) y el Mantoux fueron negativos. La radiografía de tórax fue normal. Una semana después acude de nuevo por síndrome general marcado y persistencia de la fiebre, siendo finalmente derivada al hospital para proseguir con el estudio. Fueron extraídos hemocultivos y urocultivo, con resultado negativo. Se realizó ecografía abdominal, que mostraba hepatomegalia de ecogenicidad aumentada, homogénea, sin lesiones focales, con alteración de la ecotextura hepática indicativa de afectación difusa y adenopatías a nivel epigástrico, sin dilatación de la vía biliar. La tomografía computarizada de abdomen mostró adenopatías en el tronco celíaco y la región periportal. La tomografía computarizada de tórax y colangio-RMN fueron normales. Finalmente, se realizó una biopsia hepática, en la que se puso de manifiesto una esteatohepatitis moderada con focos de necrosis hepatocelular y numerosos granulomas no caseificantes (fig. 1 A y B), compuestos por histiocitos epiteloides de tipo cuerpo extraño con citoplasma espumoso y núcleos periféricos (fig. 1C) e infiltrado inflamatorio de naturaleza linfocitaria (fig. 1D) con ausencia de necrosis central. La tinción para hongos y bacilos ácido-alcohol resistentes fue negativa. Los hallazgos histológicos, junto con los datos clínicos, de laboratorio y de imagen, fueron compatibles con el diagnóstico de SH aislada. Se inició tratamiento con corticoides a 0,5mg/kg/día (prednisona 30mg/día), y a la semana quedó apirética, con mejoría progresiva del estado general y ganancia ponderal. Al mes presentó disminución de los reactantes de fase aguda y la enzima convertidora de angiotensina, y a los 2 meses se normalizó la bioquímica hepática y se procedió a disminuir gradualmente los esteroides orales.
. B. Granuloma no caseificante de tipo sarcoideo aumentado de tamaño. C. Histiocitos epiteloides de tipo «cuerpo extraño» con citoplasma espumoso y núcleos periféricos (asteriscos). D. Infiltrado inflamatorio de naturaleza linfocitaria.")
A. Cilindro hepático con granuloma no caseificante bien delimitado (flechas). B. Granuloma no caseificante de tipo sarcoideo aumentado de tamaño. C. Histiocitos epiteloides de tipo «cuerpo extraño» con citoplasma espumoso y núcleos periféricos (asteriscos). D. Infiltrado inflamatorio de naturaleza linfocitaria.
El 95% de los pacientes con sarcoidosis sistémica tienen alguna forma de afectación hepática asociada6. La SH aislada es extremadamente rara, más frecuente en varones, con un espectro de enfermedad amplio5–7. En la mayoría de los casos (2-60%) es asintomática y se limita a alteraciones de la bioquímica hepática3,4,6. Solo el 5-30% desarrolla signos y síntomas: ictericia (rara), náuseas, vómitos, dolor abdominal (15%), hepatoesplenomegalia (2-21%), fiebre y pérdida de peso (5%)6,8,9. Menos del 1% evoluciona a cirrosis, que condiciona mal pronóstico, más si se acompaña de complicaciones7,8. El primer caso de SH con cirrosis e hipertensión portal fue reportado en 1947; desde entonces, el número de casos descritos ha ido en aumento8. La SH también puede manifestarse como colestasis crónica, intra o extrahepática1,6. La primera se debe a obstrucción de los conductos biliares por granulomas portales y periportales, y la segunda ocurre por alteración del flujo sanguíneo por compresión de los espacios porta por granulomas. En ambos casos se puede desarrollar hipertensión portal con varices esofágicas (en la colestasis intrahepática por evolución a cirrosis y en la extrahepática por aumento de la presión presinusoidal)4,5,8.
El diagnóstico de la SH es complejo, teniendo en cuenta las diferentes formas de presentación. Hallazgos de laboratorio inespecíficos, como anemia, linfopenia periférica por acumulación de linfocitos en órganos afectados, elevación de la enzima convertidora de angiotensina (alto índice de falsos positivos), hipercalcemia (por la producción de 1,25-hidroxicolecalciferol por macrófagos activados) e hipercalciuria pueden ayudar al diagnóstico6. La bioquímica hepática es anormal en el 20-50% de los casos, siendo el patrón de colestasis la alteración más frecuente3. Respecto a la evaluación radiológica de la SH, a diferencia de la sarcoidosis torácica que habitualmente muestra hallazgos característicos, ninguna prueba es útil para el diagnóstico3,4,7. No obstante, mediante ecografía se aprecia hepatomegalia en el 30-50% de los casos, con un aumento inespecífico de la ecogenicidad8. La tomografía computarizada revela hepatomegalia difusa en el 30% de los casos y solo en el 5% se aprecian lesiones nodulares múltiples, de 1-15mm, de baja atenuación y sin realce periférico tras la administración de contraste5,8,9. La SH nodular se asocia habitualmente a nódulos esplénicos y ganglios abdominales. Mediante RMN estos nódulos aparecen como lesiones hipointensas respecto al parénquima sano, sin realce tras la administración de gadolinio. El diagnóstico se confirma, apoyado por datos clínicos, de laboratorio y radiológicos, por los hallazgos histológicos de granulomas epiteloides no caseificantes junto con infiltrados de células mononucleares, lesión de hepatocitos y focos de fibrosis o cirrosis1,4,6,8. Debido al gran número y a la distribución uniforme de los granulomas, la probabilidad de falsos negativos es rara.
El tratamiento de la SH es controvertido al no haberse desarrollado grandes ensayos controlados aleatorizados5,8. Los datos son escasos y las recomendaciones actuales están basadas en casos aislados o en series de casos, sin poder extraer conclusiones definitivas debido al pequeño tamaño muestral10. Los pacientes asintomáticos no requieren tratamiento dada la rareza de complicaciones9. Tras una revisión de la literatura, la mayoría de los autores recomiendan el uso de corticoides en pacientes con SH sintomática y evidencia de colestasis o elevación de transaminasas4,10. Los corticoides han demostrado mejoría de los síntomas y de la bioquímica hepática sin alterar el curso de la enfermedad ni prevenir el desarrollo de hipertensión portal6,9. Los fármacos ahorradores de esteroides (metotrexato, azatioprina, hidroxicloroquina) serían una alternativa, pero tienen que ser evaluados con estudios a largo plazo9,10. En caso de cirrosis hepática con hipertensión portal grave, el trasplante hepático es una solución curativa, aunque se han descrito casos de recurrencia postransplante1,6,9. En nuestro caso, tras el inicio de esteroides a dosis bajas se produjo una mejoría clínica y la normalización de la bioquímica hepática con desaparición de la colestasis.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciaciónEl trabajo no recibió ninguna beca o soporte financiero.
Conflicto de interesesLos autores firmantes declaran que no existen conflictos de intereses.
