Liver regeneration is a normal response to liver injury. The aim of this study was to determine the molecular basis of liver regeneration, through an integrative analysis of high-throughput gene expression datasets.
MethodsWe identified and curated datasets pertaining to liver regeneration from the Gene Expression Omnibus, where regenerating liver tissue was compared to healthy liver samples. The key dysregulated genes and pathways were identified using Ingenuity Pathway Analysis software. There were three eligible datasets in total.
ResultsIn the early phase after hepatectomy, inflammatory pathways such as Nrf2 oxidative stress-mediated response and cytokine signaling were significantly upregulated. At peak regeneration, we discovered that cell cycle genes were predominantly expressed to promote cell proliferation. Using the Betweenness centrality algorithm, we discovered that Jun is the key central gene in liver regeneration. Calcineurin inhibitors may inhibit liver regeneration, based on predictive modeling.
ConclusionThere is a paucity of human literature in defining the molecular mechanisms of liver regeneration along a time continuum. Nonetheless, using an integrative computational analysis approach to the available high-throughput data, we determine that the oxidative stress response and cytokine signaling are key early after hepatectomy, whereas cell cycle control is important at peak regeneration. The transcription factor Jun is central to liver regeneration and a potential therapeutic target. Future studies of regeneration in humans along a time continuum are needed to better define the underlying mechanisms, and ultimately enhance care of patients with acute and chronic liver failure while awaiting transplant.
Liver regeneration occurs in response to insults that induce inflammation, cell death and injury [1], and can be categorized into acute versus chronic regeneration. Liver resection results in acute regeneration, with activation of signaling pathways, resulting ultimately in restoration of 100% of the original hepatic volume and the original liver-to-body weight ratio [2]. The liver is the only visceral organ with a tremendous capacity to regenerate in a time-limited manner with restoration of its original size. When other organs are subject to resection, there is limited local regeneration. Chronic liver regeneration occurs in response to ongoing inflammation in chronic liver diseases (viral, autoimmune, etc.). This persistent and ongoing hepatocyte proliferation in a genotoxic milieu results in the development of fibrosis and dysplasia over time [3].
The understanding of the molecular pathways underlying hepatic regeneration has been principally derived through findings from animal models [4]. Animal studies have demonstrated the dramatic gene expression changes in liver regeneration. The significant 3-fold increase in portal venous flow per hepatocyte induces various signaling changes [5], particularly early phase proteins such as urokinase plasminogen activator, Notch1, and beta-catenin [6]. Extracellular matrix remodeling is stimulated, and results in activation of Hepatocyte Growth Factor (HGF). HGF, norepinephrine, Interleukin-6 (IL-6), Tumor Necrosis Factor (TNF-alpha), serotonin, and bile acids are all found in high concentrations in the blood early after hepatectomy [7]. HGF and Epidermal Growth
Factor (EGFR) stimulate mitosis of hepatocytes [8]. In turn, the Fibroblast Growth Factor (FGFR) produced by dividing hepatocytes triggers mitosis of endothelial and stellate cells [9]. Hepatocytes then enter the cell cycle, resulting in initiation of cell proliferation, which in animals, subsides by day 7 after hepatectomy. In terms of ontology, the types of activated genes include specific transcription factors, regulators of cell cycle entry, stress and inflammatory response proteins. These proteins have a greater than 2-fold change in expression following hepatic resection [10].
Given that there is currently no bridge to liver transplantation (LT), an improved understanding of what constitutes normal liver regeneration and how to safely stimulate it has the potential for clinical impact. Cirrhotic patients with worsening liver function while awaiting LT, and those with fulminant liver failure are key patient groups who would stand to benefit from therapies generated from such an understanding. Small-for-size syndrome is associated with adverse outcomes post-LT, and would improve with stimulation of normal regeneration.
The aim of this systematic integrative analysis of all publicly available data (both human and animal) on liver regeneration is to obtain a more global understanding of the molecular basis and pathways that drive liver regeneration. Whereas most research studies tend to focus on a few key molecules, integrative analysis tools allow for an overarching picture of the drivers of a condition. Through this analysis, we will identify the key genes and pathways specific to regeneration. This would help appreciate potential therapeutic targets to drive this process as the clinical context demands it, as in acute or chronic liver failure and small-for-size syndrome.
2Methods2.1Data collection, analysis and database compilingWe retrieved all available high-throughput microarray gene expression datasets related to liver regeneration from Gene Expression Omnibus (GEO), a public functional genomics data repository containing high-throughput array data (https://www.ncbi.nlm.nih.gov/geo). We also interrogated Pubmed and ArrayExpress as per the methodology described below. A first search to identify datasets referring to human liver regeneration was performed using the following MeSH terms ((“liver regeneration”[MeSH Terms] [11] OR (“liver”[All Fields] AND “regeneration”[All Fields]) OR “liver regeneration”[All Fields]) AND high[All Fields] AND throughput[All Fields] AND “humans”[MeSH Terms]). We performed a second search to identify the datasets referring to animals, using the following search terms ((“liver regeneration”[MeSH Terms] OR (“liver”[All Fields] AND “regeneration”[All Fields]) OR “liver regeneration”[All Fields]) AND (“transcriptome”[All Fields] OR (“gene”[All Fields] AND “expression”[All Fields] AND “profile”[All Fields]) OR “gene expression profile”[All Fields])) AND “Mus musculus”[porgn] OR “Rattus norvegicus” [porgn] OR “Oryctolagus cuniculus”[porgn]. We included all high-throughput gene expression profiling datasets comparing liver regeneration at different time points to normal liver tissue (non-regenerating) at baseline as control in humans and mice. These datasets on GEO were analyzed using GEO2R (https://www.ncbi.nlm.nih.gov/geo/info/geo2r.html), a web tool available on the portal, to identify genes differentially expressed between samples of liver regeneration and normal liver. GEO2R compares original submitter-supplied processed data tables using the GEOquery [12] and limma package [13] from the Bioconductor project. Following the instructions available online, we collected all the dysregulated genes with an adjusted p-value p<0.05, and an expression fold-change value below 0.5 or above 1.5. The study workflow is illustrated in Fig. 1.
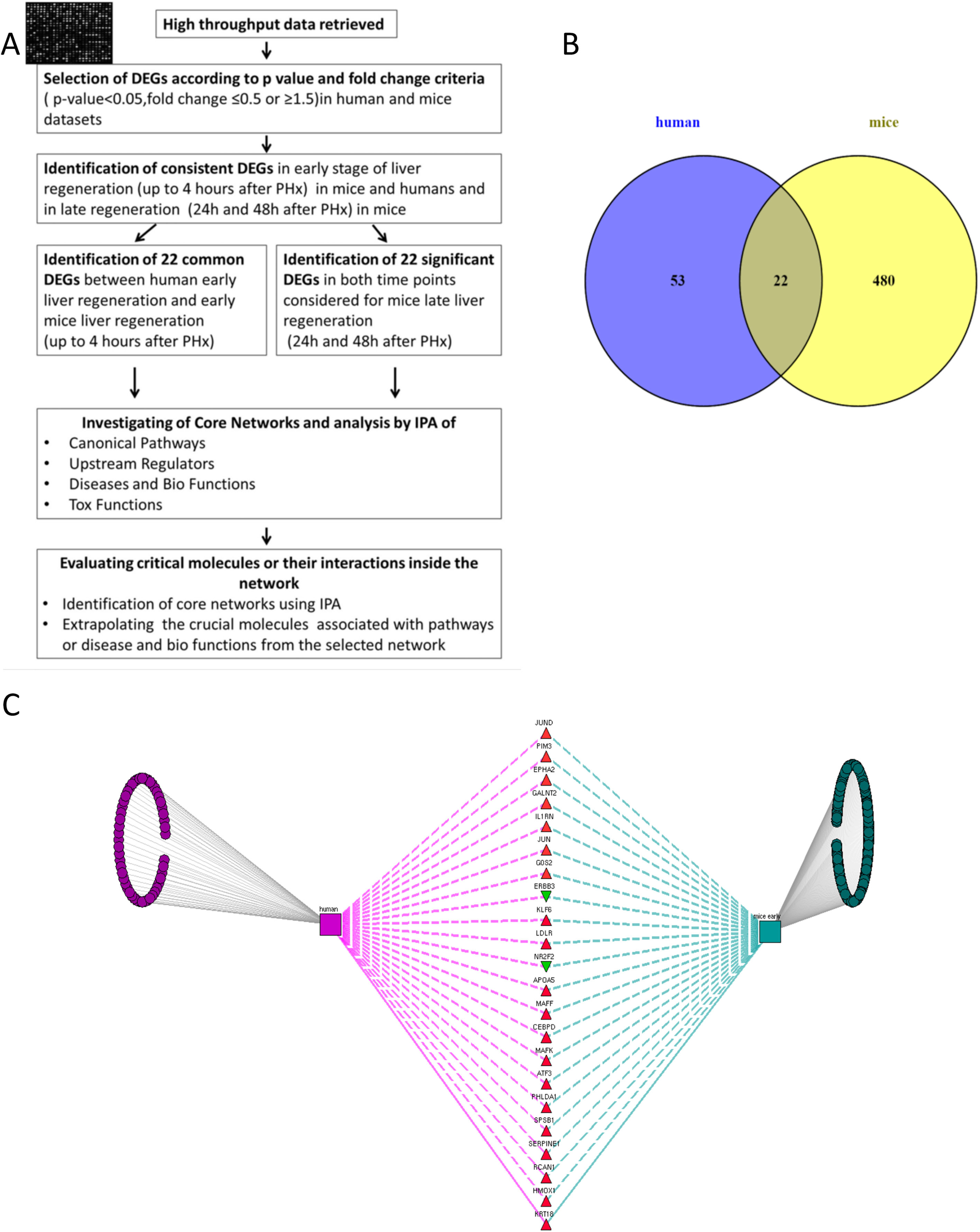
 Flow chart detailing the steps in our study, from data curation to final generation of networks based on protein-protein interactions. (B) Venn Diagram illustrating the distinct and overlapping dysregulated genes between human and mouse datasets. There were 75 dysregulated genes in humans, 502 dysregulated genes in mice, and 22 shared genes between human and mice (list of genes reported in Supplementary file 1). (C) Network analysis demonstrates the genes common to human and mouse early liver regeneration in the center, with the red triangles depicting up-modulated genes, and the green triangles depicting the down-modulated genes.")
(A) Flow chart detailing the steps in our study, from data curation to final generation of networks based on protein-protein interactions. (B) Venn Diagram illustrating the distinct and overlapping dysregulated genes between human and mouse datasets. There were 75 dysregulated genes in humans, 502 dysregulated genes in mice, and 22 shared genes between human and mice (list of genes reported in Supplementary file 1). (C) Network analysis demonstrates the genes common to human and mouse early liver regeneration in the center, with the red triangles depicting up-modulated genes, and the green triangles depicting the down-modulated genes.
We used 22 deregulated genes found as commonly modulated between human early regeneration and mice for the core network analysis. An overview of the study flow for the network analysis is illustrated in Fig. 1. To investigate the interactions between the modulated genes found in common between mice and human regenerative liver, we used Ingenuity pathway analysis (IPA Ingenuity® Systems, www.ingenuity.com). IPA identifies network interactions and pathway interactions between genes based on an extensive manually curated database of published gene interactions. We uploaded the gene symbol and the associated expression value from the microarray data analyzed with GEO2R into IPA. These genes, called focus genes, were overlaid onto a global molecular network based on Ingenuity knowledge database. IPA includes a large repository of gene–phenotype associations, molecular interactions, chemical knowledge, and regulatory events, manually curated from scientific publications. The networks based on these focus genes were algorithmically created based on their direct or indirect interactions. Scores, calculated on Fisher's exact test by IPA, were obtained in order to rank networks based on their relevance to the genes initially uploaded. The score evaluates the number of focus genes from our original dataset in the network, and the size of the network to approximate how relevant this network is to the original list of focus genes. The network is then shown as a graph representing the molecular relationships/interactions as an edge (line) between genes or gene products (nodes). The connectivity of these nodes representing the genes is based on the data collected in the IPA knowledge base. The node color indicates an up-modulation (red) or down-modulation (green). Nodes are displayed using various shapes to represent the functional role or class of gene product (i.e. kinase, transcription regulator, enzyme). Edges are displayed with various colors or labels to better describe the nature of the relationship between the nodes. Common deregulated genes were mapped onto the core networks to explore their connection to biological function or disease affecting the liver. The upstream regulators significantly associated with our list of Differentially Expressed Genes (DEGs) at different time points were obtained by IPA Ingenuity's knowledge base, as well as the canonical pathways significantly associated with the genes in the input dataset. The right-tailed Fisher's exact test was used to calculate a p-value determining the probability that the association or overlap between the genes listed in the dataset and a given pathway's neighborhood was due to chance alone.
Based on the modulation of the DEGs, IPA is able to calculate a z score, defined as a statistical measure of correlation between relationship direction and a given set of modulated genes, as a value to measure “the non-randomness” of directionality. Z score <−2 or > +2 is considered significant. Negative z score <−2 predicts an inhibition with high confidence and a positive score >+2 predicts activation with strong confidence. Outside these values, the prediction of activation or inhibition is less confident. In some cases, the z score cannot be calculated, if there is not enough information stored on IPA knowledge base, in which case there is no available prediction.
2.3Hepatic regeneration protein–protein interaction network analysisWe merged and overlaid core networks with related “functions and diseases” to determine genes associated with specific hepatic biological and pathological processes according to the IPA knowledge base. We used the MAP (Molecular Activity Predictor) tool to predict the crosstalk relationship among our genes and their interactors based on their modulation for the networks considered. The drug database was overlaid with Core Network #1 in early regeneration to identify the possible effect of calcineurin inhibitors on liver regeneration.
For later regeneration at 48h, Core Network #3 was overlaid with canonical pathways and the enriched pathway, and their relationship with the network is shown in Supplementary Fig. 5A.
2.4Calculation of centrality in protein–protein interaction networksFor all the 22 DEGs in early and late regeneration, we retrieved the known interactors using the Integrated Interactions Database (IID at http://ophid.utoronto.ca/iid) [14] version 2016-03, selecting only data in humans, mice and liver tissue. Betweenness centrality was calculated using the betweenness function in the igraph package version 1.0.1 [15], in R version 3.3.1 (https://www.R-project.org/).
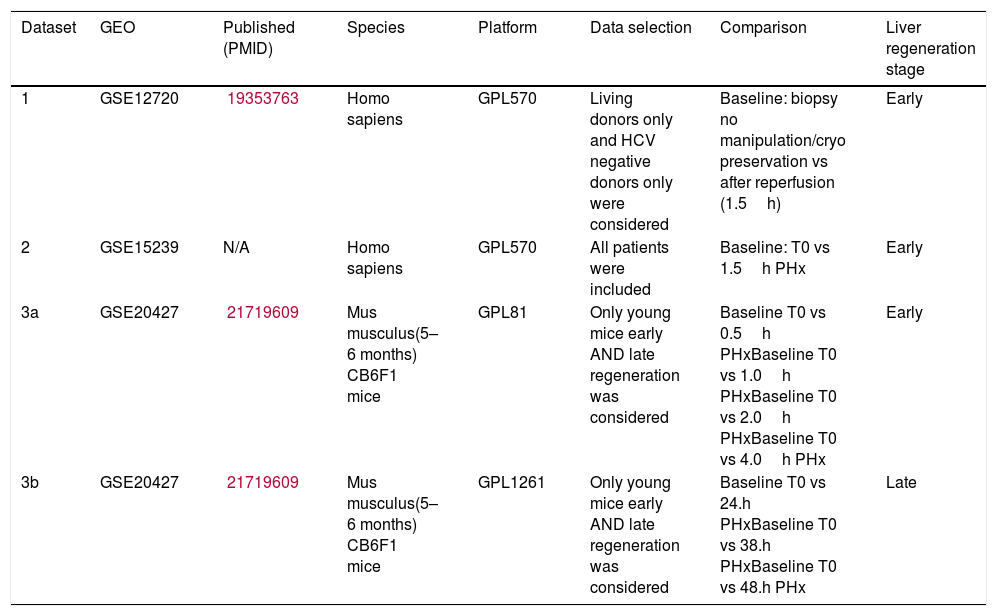
3Results3.1Datasets pertaining to liver regenerationWe retrieved 7 datasets for humans and 16 datasets for mice from GEO. However, most patient datasets were not investigating liver regeneration at all, and most mouse datasets did not include different time points. The final selected studies are listed in Table 1, one of which is a mouse study establishing gene expression data for both early and late liver regeneration. The other two datasets were human: one was obtained from 8 living donor liver grafts following reperfusion, the other was from 3 patients of varying ages (1.5, 42 and 81 years old) post-hepatectomy.
List of datasets included in our integrative analysis.
| Dataset | GEO | Published (PMID) | Species | Platform | Data selection | Comparison | Liver regeneration stage |
|---|---|---|---|---|---|---|---|
| 1 | GSE12720 | 19353763 | Homo sapiens | GPL570 | Living donors only and HCV negative donors only were considered | Baseline: biopsy no manipulation/cryo preservation vs after reperfusion (1.5h) | Early |
| 2 | GSE15239 | N/A | Homo sapiens | GPL570 | All patients were included | Baseline: T0 vs 1.5h PHx | Early |
| 3a | GSE20427 | 21719609 | Mus musculus(5–6 months) CB6F1 mice | GPL81 | Only young mice early AND late regeneration was considered | Baseline T0 vs 0.5h PHxBaseline T0 vs 1.0h PHxBaseline T0 vs 2.0h PHxBaseline T0 vs 4.0h PHx | Early |
| 3b | GSE20427 | 21719609 | Mus musculus(5–6 months) CB6F1 mice | GPL1261 | Only young mice early AND late regeneration was considered | Baseline T0 vs 24.h PHxBaseline T0 vs 38.h PHxBaseline T0 vs 48.h PHx | Late |
Identification of the DEGs early post-hepatectomy that were overlapping in humans and mice, as opposed to distinct to each, was performed using a Venn diagram as shown in Fig. 1. We focused on these overlapping genes for further analysis, given that these overlapping genes demonstrated consistency with respect to their involvement in early liver regeneration. The 22 genes in common, and their modulation are listed in Fig. 1.
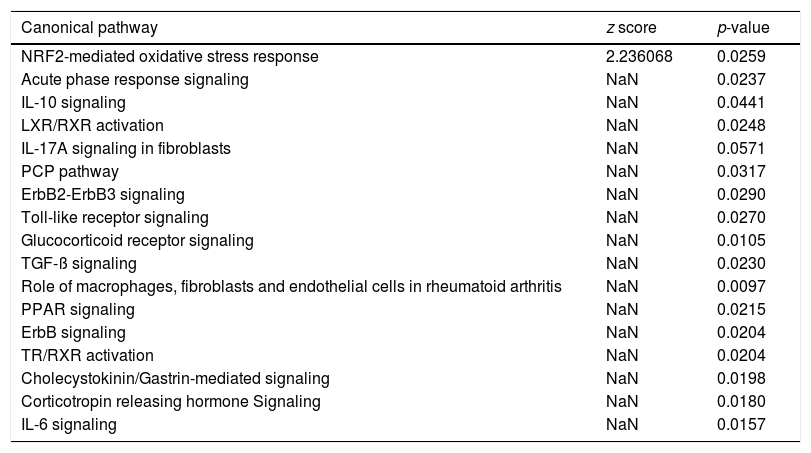
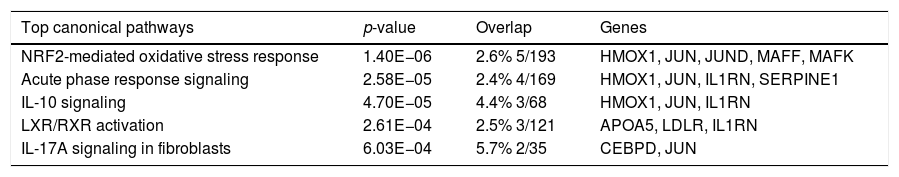
Canonical pathway analysis revealed the key signaling cascades in which these genes are involved, as listed in Table 2, highlighting the importance of inflammation as a driver of the hepatic regenerative process. The NRF2-mediated Oxidative Stress Response was not only the most significantly enriched pathway in early regeneration, but also predicted to be activated (z score value >2) based on the modulation status of the 22 DEGs. Cytokine-associated pathways such as IL-10, IL-17 and IL-6, previously highlighted in the regeneration literature [16], were also significantly associated with our gene list. Table 3 reports the key genes among the 22 involved in the most significant pathways, along with their degree of involvement according to percentage of the total number of genes in the pathway.
Canonical pathway analysis of the genes common to human and mouse regeneration resulted in a list of significantly enriched pathways (p-value <0.05), the activation status of each pathways is based on the z score (−2<z score=inhibition-high confidence, −2<z score<0=inhibition-less confidence; 0>z score<+2=activation-less confidence; z score<+2=strong activation-high confidence; NaN=no prediction possible).
| Canonical pathway | z score | p-value |
|---|---|---|
| NRF2-mediated oxidative stress response | 2.236068 | 0.0259 |
| Acute phase response signaling | NaN | 0.0237 |
| IL-10 signaling | NaN | 0.0441 |
| LXR/RXR activation | NaN | 0.0248 |
| IL-17A signaling in fibroblasts | NaN | 0.0571 |
| PCP pathway | NaN | 0.0317 |
| ErbB2-ErbB3 signaling | NaN | 0.0290 |
| Toll-like receptor signaling | NaN | 0.0270 |
| Glucocorticoid receptor signaling | NaN | 0.0105 |
| TGF-ß signaling | NaN | 0.0230 |
| Role of macrophages, fibroblasts and endothelial cells in rheumatoid arthritis | NaN | 0.0097 |
| PPAR signaling | NaN | 0.0215 |
| ErbB signaling | NaN | 0.0204 |
| TR/RXR activation | NaN | 0.0204 |
| Cholecystokinin/Gastrin-mediated signaling | NaN | 0.0198 |
| Corticotropin releasing hormone Signaling | NaN | 0.0180 |
| IL-6 signaling | NaN | 0.0157 |
The top canonical pathways associated with genes common to human and mouse regeneration, with p-value and gene overlap expressed in percentage and ratio of number of genes to the total elements of each pathway.
| Top canonical pathways | p-value | Overlap | Genes |
|---|---|---|---|
| NRF2-mediated oxidative stress response | 1.40E−06 | 2.6% 5/193 | HMOX1, JUN, JUND, MAFF, MAFK |
| Acute phase response signaling | 2.58E−05 | 2.4% 4/169 | HMOX1, JUN, IL1RN, SERPINE1 |
| IL-10 signaling | 4.70E−05 | 4.4% 3/68 | HMOX1, JUN, IL1RN |
| LXR/RXR activation | 2.61E−04 | 2.5% 3/121 | APOA5, LDLR, IL1RN |
| IL-17A signaling in fibroblasts | 6.03E−04 | 5.7% 2/35 | CEBPD, JUN |
Additional analysis of upstream activators of the identified pathways using IPA revealed 16 molecules including 5 cytokines (TNF, IL-1B, IFN-G, IL-6, IL-1A), 3 transcriptional factors (EGR1, PDX1, NUPR1), 4 growth factors (Epidermal Growth Factor (EGF), growth hormone 1 (GH1), angiopoietin-related growth factor (AGF), hepatocyte growth factor (HGF)) and 4 kinases (p38,ERK, JNK, MAP2K1/2). The impact of the significant growth factors on the key genes and biological processes in liver regeneration are illustrated in Fig. 2.
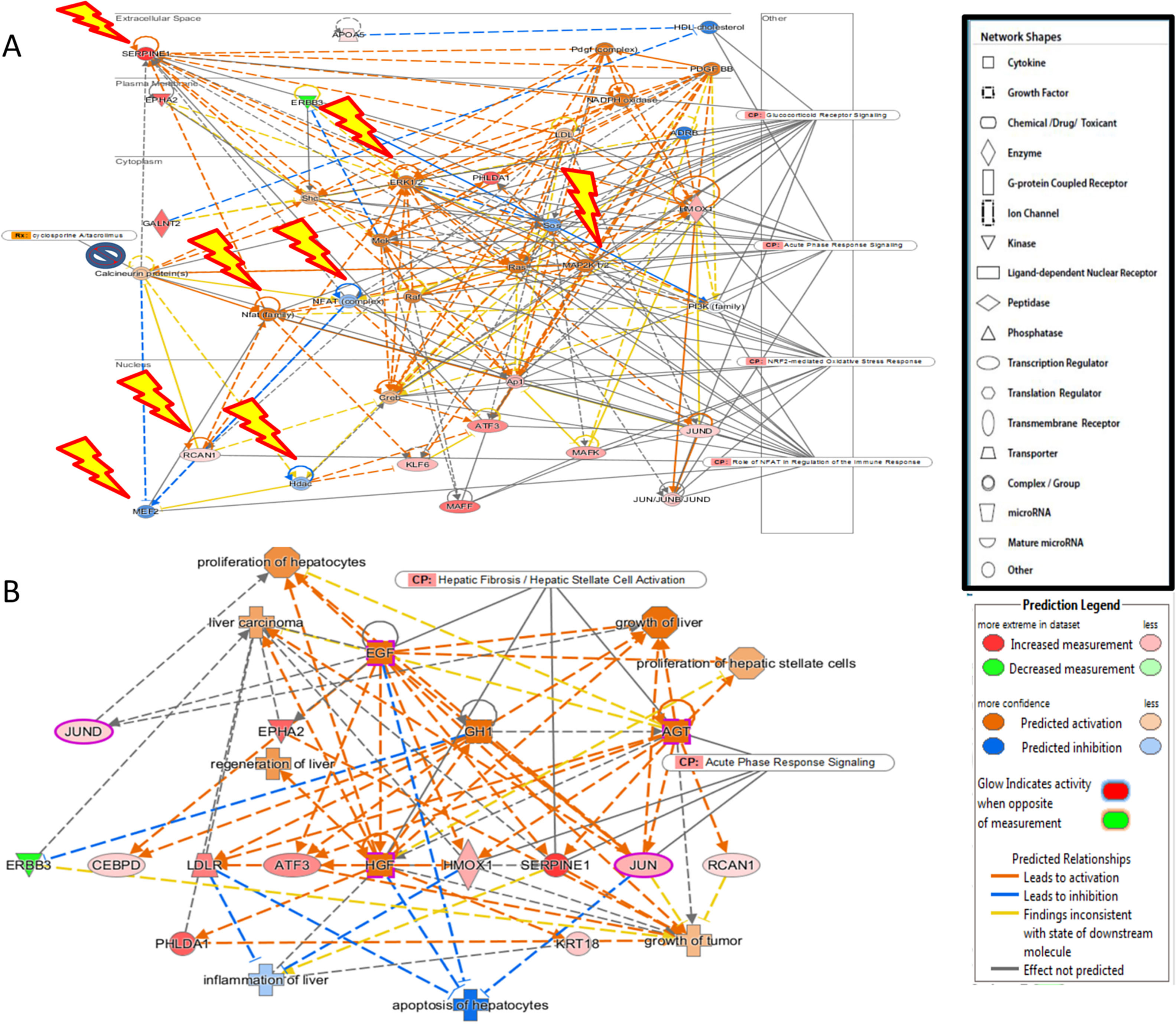
 The 4 significant growth factors (Epidermal Growth Factor (EGF), growth hormone 1 (GH1), angiopoietin-related growth factor (AGF), Hepatocyte growth factor (HGF)) and their impact on the key genes and biological processes in liver regeneration are illustrated. The biological processes (represented by diamond) as elucidated by the Ingenuity Pathway Analysis software include hepatocyte proliferation, liver regeneration, hepatic stellate cell proliferation, but can also promote carcinogenesis (pathological consequences are represented by cross). (B) Predicted effect of calcineurin inhibitors tacrolimus and cyclosporine on Liver REGENERATION. Based on knowledge of protein-protein interactions with the Calcineurin protein(s), depicted in Core Network#1 we can predict the inhibitory effect of calcineurin inhibitors on the downstream pathways in the acute phase after hepatectomy including NRF2-mediated oxidative stress response, glucocorticoid receptor signaling, and acute phase response signaling.")
(A) The 4 significant growth factors (Epidermal Growth Factor (EGF), growth hormone 1 (GH1), angiopoietin-related growth factor (AGF), Hepatocyte growth factor (HGF)) and their impact on the key genes and biological processes in liver regeneration are illustrated. The biological processes (represented by diamond) as elucidated by the Ingenuity Pathway Analysis software include hepatocyte proliferation, liver regeneration, hepatic stellate cell proliferation, but can also promote carcinogenesis (pathological consequences are represented by cross). (B) Predicted effect of calcineurin inhibitors tacrolimus and cyclosporine on Liver REGENERATION. Based on knowledge of protein-protein interactions with the Calcineurin protein(s), depicted in Core Network#1 we can predict the inhibitory effect of calcineurin inhibitors on the downstream pathways in the acute phase after hepatectomy including NRF2-mediated oxidative stress response, glucocorticoid receptor signaling, and acute phase response signaling.
The predicted impact of calcineurin inhibitors is illustrated in Fig. 2. Integration of Core Network #1 and drugs targeting calcineurin suggests that the most commonly used immunosuppressants in liver transplantation adversely affect liver regeneration.
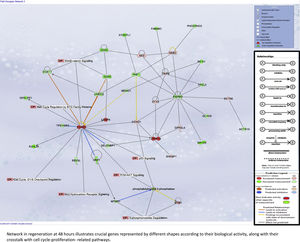
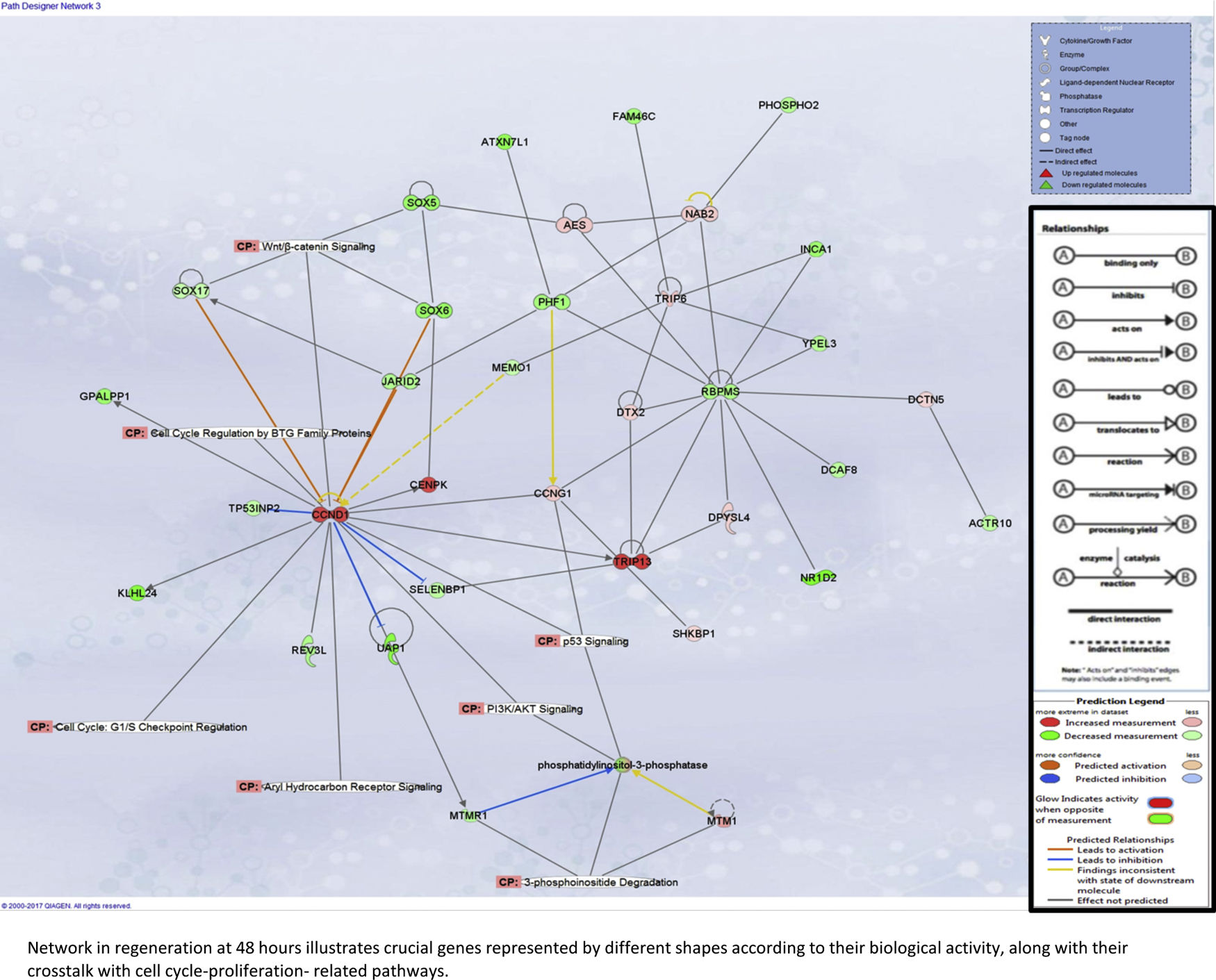
Liver regeneration at the later time point of 48h appears to be driven by cell cycle-related genes, as illustrated in Supplementary Fig. 5A.
4DiscussionThe liver is unique in being capable of rapid regeneration in response to various stimuli, such as injury and hepatectomy. Although it is a key physiological process shown to be well-orchestrated based on in vivo data, liver regeneration in humans along a time continuum remains incompletely understood. In this integrative analysis of high-throughput datasets, we systematically deciphered the molecular basis of liver regeneration by studying and analyzing all publicly available high-throughput gene expression datasets. We discovered that the high-throughput data in liver regeneration has been limited to the early phase of regeneration. Particularly, the data in humans has been restricted to the first 1.5h after hepatectomy, which may be more representative of acute stress than a regenerative response. Given that liver regeneration in humans is a process that takes up to 12 weeks to complete following hepatectomy for living donation, with a percentage reconstitution of 80%±13% [17], this focus on the early period after hepatectomy hinders our understanding of human liver regeneration along a time continuum. The mouse data was more representative of changes over a time continuum, and there were certainly overlapping genes between humans and mice in the early phase based on integration of the publicly available data.
We discovered that the early phase after hepatectomy is dominated by inflammation-associated pathways, particularly nuclear factor erythroid 2-related factor 2 (Nrf2) oxidative stress response [18]. Nrf2 is a transcription factor that plays a key role in cytoprotection in the face of ongoing injury from reactive oxygen species and radicals induced by viruses, toxins, and lipid accumulation in the liver [19]. The crucial role of Nrf2 in activating liver regeneration has been supported by in vivo data [18]. Increased oxidative stress leads to insulin/Insulin Growth Factor resistance in Nrf2-deficient hepatocytes, which in turn prevents efficient regeneration [20]. It has been shown that Nrf2 protects the liver from toxin-mediated damage and decreases fibrogenesis, which is partly due to target genes regulated by Nrf2 in hepatocytes [21]. The IL-6[22] and IL-17[16] -associated pathways were also key to liver regeneration, as previously confirmed in the literature. We also identified the IL-10 pathway as a top canonical pathway, which concords with previous studies demonstrating that the anti-inflammatory IL-10 inhibits liver regeneration [23]. The role of inflammatory cells in regeneration has previously been highlighted in a systems analysis of non-parenchymal cell modulation in liver repair, where a balance of inflammatory signals and growth factors was reported [24].
The LXR/Retinoid × Receptor pathway was among the top canonical pathways identified, and has clearly been shown to play a role in liver regeneration in vivo [25–28]. In fact, retinoid stores regulate cell cycle gene expression, and stimulate liver regeneration [27,28]. We identified immediate early genes such as JUN and MYC as being central to the network in early regeneration after two-thirds hepatectomy, which is compatible with the literature[29].
Our assessment of the genes and pathways in peak liver regeneration was limited to a single mouse dataset due to the paucity of high-throughput data. Cell cycle genes were the most clearly implicated in late regeneration, along with Wnt/B-catenin and PI3K/Akt/mTOR pathways.
A key finding in our study was the discovery of that the transcription factor JUN is a gene central to regeneration. This indicates that the presence of Jun in the protein-protein interaction networks is essential to driving hepatic regeneration. The importance of this critical growth-related gene has been previously demonstrated in stimulating hepatocyte proliferation [30], with a liver-specific gene deletion in mice resulting in impaired liver regeneration post-hepatectomy [31]. Additionally, JUN has been shown to be critical in the early regenerative response to liver injury [32,33] and small-for-size syndrome in vivo [34].
Our analysis of biological processes revealed an overlap of genes involved in liver regeneration and hepatic carcinogenesis. This has also been verified in the literature, where hierarchical clustering of liver regeneration and hepatocellular carcinoma-related demonstrated overlapping involvement in the cell cycle and cell death. Rapid proliferation in the context of injury carries the risk of accumulation of mutations [35]. Chronic liver regeneration in response to genotoxic liver injury also is associated with development of dysplasia [36,37].
Given that living donor liver transplant recipients have liver regeneration in the first 3 months after transplant, we wished to investigate the potential effect of calcineurin inhibitors (the most commonly used immunosuppressants after liver transplant) on the pathways upregulated post-hepatectomy using the IPA software. Specifically, we situated the effect of the calcineurin proteins in these pathways early after hepatectomy, and saw that Nrf2-mediated oxidative stress response, the acute phase response and glucocorticoid signaling [38] would all be potentially impaired with calcineurin inhibition. It is possible that calcineurin inhibitors prevent liver regeneration in response to genotoxic injury such as viruses and lipid accumulation, and may be a link to the hastened progression of fibrosis following liver transplantation [39]. However, there have previously been reports of increased mitotic activity in the presence of tacrolimus [40,41] and cyclosporine [42], although regenerative capacity and the resulting liver volume have not been assessed. Therefore, the exact impact of calcineurin inhibitors on hepatic regeneration remains to be determined. The only alternative baseline immunosuppressant is sirolimus, which as an mTOR inhibitor is an antiproliferative agent and is contraindicated in the first month post-transplant due to the heightened risk of fatal hepatic artery thrombosis.
The principal limitation of this integrative analysis is the paucity of publicly available high-throughput gene expression data. Given that our understanding of human regeneration is limited to the early phase post-hepatectomy or living donor liver transplant, the data is more influenced by acute stress and inflammatory processes such as ischemia-reperfusion injury. We did find animal data pertaining to what corresponds to their peak liver regeneration time point, however there was no such corresponding human data available. There was no high-throughput data available on termination of liver regeneration, therefore this phase could not be studied. It has been shown that the liver has an inherent hepatostat, regulating the organ size achieved [43]. However, the mechanisms underlying termination of regeneration are not well understood due to paucity of data. Nonetheless, limited data does indicate the key role of extracellular matrix-driven intracellular signaling involving proteins such as integrin-linked kinase, Glypican 3, C/EBPα, and HNF4α [43].
The literature on hepatic regeneration has been extensively influenced by animal data, and it is doubtful that this is an accurate representation of human regeneration, especially given that it occurs over a longer time frame. Nonetheless, we did uncover a commonality in genes expressed early after hepatectomy. Another important aspect is the likely contribution of different liver cell subsets such as hepatocytes, macrophages, and other immune cells to the regenerative process. The datasets obtained from the public literature represent gene expression in whole liver tissue, and do not enhance our understanding of how individual cell types contribute to hepatic regeneration.
In summary, liver regeneration is a key biological process that occurs in various contexts, from chronic injury in liver disease, to partial hepatectomy performed for various indications, to living donor liver transplantation. Our systematic review and integrative analysis of high-throughput gene expression data in liver regeneration provides a picture of the processes involved early after hepatectomy and at peak liver regeneration. Inflammatory pathways predominate the early period after hepatectomy, and cell cycle-related genes drive regeneration at its peak, at least in animals. Additionally, JUN is a transcription factor that is central to regeneration, and is thus critical for this physiological process. Based on our centrality analysis of high-throughput data and protein–protein interactions, without the presence of Jun, the regenerative process would fail to occur. Validation of the central role of Jun is necessary to determine its importance as a therapeutic target to stimulate liver regeneration. Nonetheless, data to better understand the molecular basis of liver regeneration in humans on a time continuum is lacking. The effect of calcineurin inhibitors on liver regeneration may play a role in hastened fibrosis post-liver transplantation. Overall, our study strongly supports the need for more extensive data on liver regeneration, with the potential to significantly enhance the care of patients with acute and chronic liver failure as well as small-for-size syndrome.
AbbreviationsDEG differential expressed genes epidermal growth factor fibroblast growth factor gene expression omnibus growth hormone 1 hepatocyte growth factor interleukin 6 ingenuity pathway analysis liver transplant tumor necrosis factor alpha
MB study design, and writing of manuscript. CB, EP and MA; data collection, analysis and compiling. AH, SM, JF, IM and MB input into study design and final manuscript.
Conflict of interestThere is no conflict of interest concerning this study.