





P-462 - RESECCIÓN DE TUMOR FIBROSO SOLITARIO INGUINAL RECIDIVADO
Hospital de Basurto-Osakidetza, Bilbao.
Introducción: El tumor fibroso solitario (TFS) es una neoplasia rara, de origen mesenquimal y potencial maligno. Algunos autores lo clasifican como sarcoma de tejidos blandos (STB). Su localización es habitualmente pleural, siendo la inguinal extremadamente infrecuente. Presentamos el caso de una paciente con TFS inguinal recidivado y su manejo.
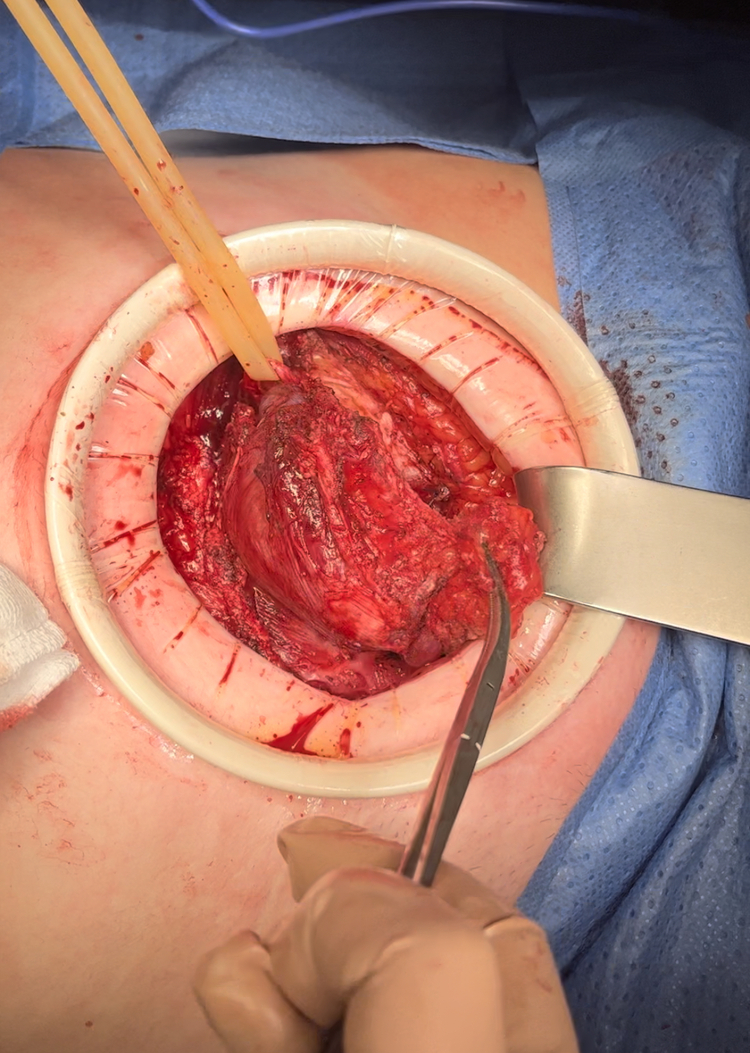
Caso clínico: Mujer de 53 años intervenida en 2011 de hernia inguinal izquierda (técnica Lichtenstein) con hallazgo intraoperatorio de una tumoración de 5 cm que se extirpó e histológicamente compatible con neurofibroma. Consulta años después por tumoración inguinal izquierda de crecimiento lento progresivo, dolorosa, dura, irreductible. La TC y RM muestran dos tumoraciones de 68 × 49 × 59 mm y 38 × 20 × 27 mm, contactando con vasos ilíacos, musculatura oblicua y vejiga pero sin infiltración ni diseminación. Se realiza biopsia ecoguiada con resultado de TFS. La paciente es intervenida mediante incisión suprapúbica, accediendo al espacio preperitoneal. Se objetiva un tumor de 8cm bien delimitado desde cresta ilíaca izquierda hasta pubis y otro de 4cm bajo la vena ilíaca. Se realiza resección en bloque incluyendo la musculatura oblicua, quedando parcialmente desprovista de pared abdominal anterior. Se repara mediante malla de polipropileno fijada a pubis, ligamento de Cooper, cintilla iliopubiana y musculatura oblicua. La evolución posoperatoria fue satisfactoria, siendo dada de alta al 6º día. El análisis histológico muestra un TFS con dos nódulos (Ki67 20% y 7%) y márgenes libres salvo afectación focal del margen vascular. Según la clasificación de 2020 de la OMS correspondería al grupo de riesgo metastásico intermedio-bajo. Valorada en comité multidisciplinar se decide tratamiento radioterápico adyuvante. No presenta recidivas hasta el momento.
Discusión: El TFS es una neoplasia fibroblástica de origen mesenquimatoso con potencial agresividad local y metastásica (2-3,7% de STB). Presenta una incidencia < 1/1.000.000, igual en hombres y mujeres, mayores de 60 años con predilección por membranas serosas (50% pleural, 20% retroperitoneal o pélvica). Generalmente son asintomáticos y suelen hallarse incidentalmente en pruebas de imagen. El diagnóstico se basa en la biopsia. Histológicamente crea dudas diagnósticas cuando no presenta clara malignidad. Son positivos para CD34 (90%), CD99, bcl-2, vimentina y reordenamiento genético NAB2-STAT6. El único tratamiento curativo es quirúrgico: la resección R0 mejora la supervivencia y disminuye la recidiva. Actualmente no está indicada la resección de órganos adyacentes salvo infiltración. La mayoría se comportan de manera indolente. El 10-15% presenta comportamiento agresivo (mayor riesgo de recidiva metastásica que local). Los factores de riesgo de recidiva son: resección incompleta, enfermedad metastásica, tamaño, índice mitótico, necrosis tumoral y edad avanzada. A diferencia de otros sarcomas, tienen una supervivencia global a 5 años del 80% y la tasa de recidiva local a los 5 años es baja (5-10%). Las metástasis son menos frecuentes, aunque incrementan hasta un 17% si cumplen factores riesgo. Pueden ocurrir recidivas tardías (10-40% hasta 20 años) por lo que el seguimiento a largo plazo está indicado. Si son resecables, la supervivencia es prolongada. El tratamiento adyuvante aún no está bien establecido o definido aunque pueden administrarse quimioterapia y/o radioterapia en resecciones incompletas o recurrencias.

