



La epigenética se define como el estudio de los mecanismos que regulan la expresión génica sin modificar la secuencia de ADN, siendo entre ellos el más conocido la metilación del ADN. La esclerosis múltiple (EM) es una enfermedad de etiología no del todo conocida, en la que se plantea que la participación de factores ambientales sobre individuos con una determinada predisposición genética, pueden resultar claves para el desarrollo de la enfermedad. Es en esta intersección entre la predisposición genética y los factores ambientales donde la metilación del ADN puede desempeñar un papel patogénico.
DesarrolloRealizamos una revisión bibliográfica de los efectos que los factores de riesgo ambiental para el desarrollo de EM pueden ejercer sobre los distintos mecanismos epigenéticos, así como la implicación que presentan dichas modificaciones en el desarrollo de la enfermedad.
ConclusiónEl conocimiento de las modificaciones epigenéticas involucradas en la patogenia de la EM abre una nueva vía de investigación para la identificación de potenciales biomarcadores, así como para la búsqueda de nuevas dianas terapéuticas.
Epigenetics is defined as the study of the mechanisms that regulate gene expression without altering the underlying DNA sequence. The best known is DNA methylation. Multiple Sclerosis (MS) is a disease with no entirely known etiology, in which it is stated that the involvement of environmental factors on people with a genetic predisposition, may be key to the development of the disease. It is at this intersection between genetic predisposition and environmental factors where
DNA methylation may play a pathogenic role. Development: A literature review of the effects of environmental risk factors for the development of MS can have on the different epigenetic mechanisms as well as the implication that such changes have on the development of the disease.
ConclusionKnowledge of epigenetic modifications involved in the pathogenesis of MS, opens a new avenue of research for identification of potential biomarkers, as well as finding new therapeutic targets.
La primera aparición del término epigenética en la literatura data de mediados del siglo XX (Conrad Waddington, 1905-1975)1.Sin embargo, ha sido en los últimos años cuando se ha convertido en uno de los campos de investigación emergentes, presentándose como una prometedora fuente de conocimientos, especialmente en el ámbito de la medicina.
La epigenética se define como el estudio de los mecanismos que regulan la expresión génica sin modificar la secuencia del ácido desoxirribonucleico (ADN). Esta disciplina representa un puente entre las influencias genéticas y ambientales en el desarrollo de un fenotipo. Los cambios epigenéticos permiten que unos genes se expresen o no, en función de condiciones exteriores, y son esenciales en la diferenciación celular y tisular que tiene lugar durante el desarrollo embrionario, y también, en los organismos adultos. De esta forma nuestras células sufren cambios epigenéticos durante toda la vida. De hecho, gemelos idénticos, con igual carga genética, acumulan diferentes patrones epigenéticos dependiendo de los factores ambientales a los que se vean sometidos, como por ejemplo, el tabaco, la alimentación, o el ejercicio2. Y esto, se traduce en diferencias observables en el fenotipo de ambos gemelos, ya sea un comportamiento distinto o un riesgo diferente de padecer enfermedades3.
Los principales mecanismos epigenéticos comprenden la metilación del ADN, la modificación de las histonas y la acción de los ARN no codificantes. De ellos, el más conocido y sobre el que más estudios se han realizado en cuanto a su relación con el desarrollo de enfermedades, es la metilación del ADN, siendo el mecanismo sobre el que centramos nuestra revisión.
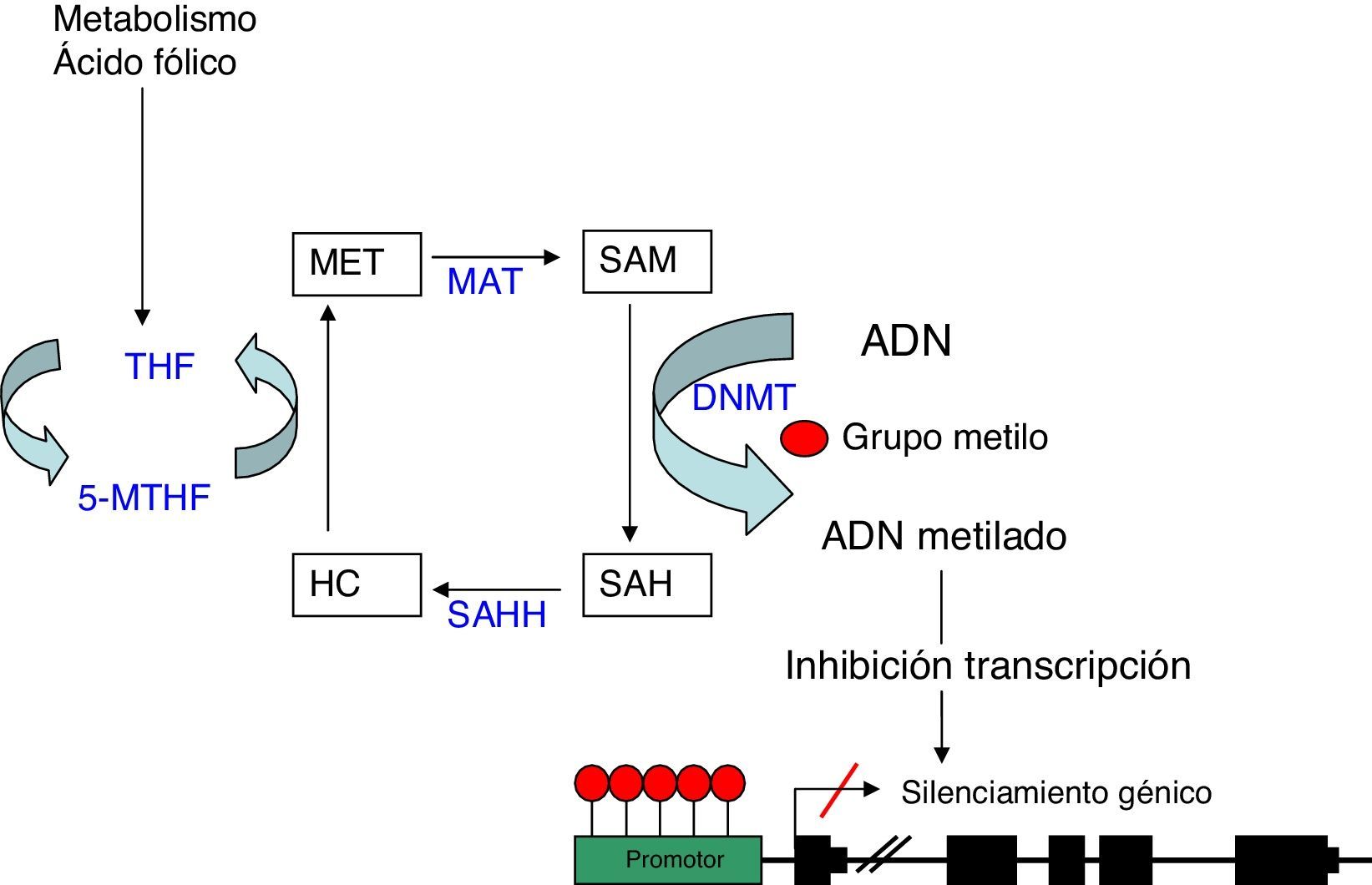
Metilación del ADNLa metilación del ADN consiste en la unión de un grupo metilo a un residuo de citosina en la cadena de nucleótidos del ADN. Esta unión se produce en los dinucleótidos citosina-guanina (CpG), los cuales, se agrupan en el genoma constituyendo las llamadas islas CpG. Estas son especialmente abundantes en las regiones promotoras de los genes y en otras zonas reguladoras. La metilación es llevada a cabo por las ADN metiltransferasas (DNMT) que catalizan la transferencia de un grupo metilo de la S-adenosil-L-metionina (SAM) al carbono 5 de la citosina4. Este proceso se puede llevar a cabo según 2 modelos diferentes: la aparición de un patrón de metilación «de novo» catalizado por las enzimas DNMT3a y DNMT3b5, o bien por el mantenimiento de un patrón de metilación en los sucesivos ciclos de replicación celular realizado por la DNMT1. Esta metilación tiene lugar durante la replicación del ADN, de manera que cuando una secuencia CpG adquiere un patrón de metilación determinado, esta modificación se hace estable, lo cual permite que sea heredada durante la duplicación del ADN y se mantenga así en las células hijas6.
La hipermetilación de las islas CpG en la región promotora de los genes es habitualmente un mecanismo de represión génica al inhibir la transcripción. Esta inhibición se realiza básicamente a través de 2 procesos, uno de manera directa, al impedir la unión de los factores de transcripción que contengan sitios de reconocimiento para las CpG metiladas. Y el otro, de manera indirecta, al bloquear el acceso a los elementos reguladores, necesarios para la unión de los factores de transcripción, mediante la adhesión de complejos proteicos denominados «methyl binding domain» (MBD) que se unen a las regiones CpG metiladas7.
Como comentábamos anteriormente, el donador del grupo metilo es la molécula SAM, la cual, una vez pierde dicho grupo metilo, se convierte en S-adenosil- homocisteína (SAH). Esta molécula se hidroliza a homocisteína, para posteriormente ser remetilada a metionina gracias al cofactor 5metilentetrahidrofolato (5MTHF). Finalmente la metionina se convierte nuevamente en una molécula SAM por acción de la metionina adenosiltrasferasa (MAT) (fig. 1). El potencial de metilación del ADN va a depender del cociente del nivel de SAM entre SAH en sangre. A mayor cociente, mayor potencial de metilación8. Por lo tanto, cabe inferir, que para el proceso de metilación del ADN, es de vital importancia el correcto metabolismo de la homocisteína y la metionina, así como el de las diferentes enzimas que participan en esta ruta metabólica y de otras sustancias como el ácido fólico y la vitamina B129.

Metilación del ADN: representación esquemática de la ruta metabólica implicada en la metilación del ADN. DNMT: DNA metiltransferasa; HC: homocisteina; MAT: metionina adenosiltrasferasa; Met: metionina; SAH: S-adenosilhomocisteina; SAHH: sadenosilhomocisteina hidrolasa; SAM: S-adenosilmetionina; THF: tetrahidrofolato; 5-MTHF: 5-metilentetrahidrofolato.
La implicación de las alteraciones de los mecanismos epigenéticos en el desarrollo de enfermedades se ha situado en los últimos años como una vía de investigación emergente, obteniéndose resultados positivos en distintas patologías, especialmente en el campo de la oncología. El primer tumor en relacionarse con los mecanismos de regulación epigenética fue el cáncer colorrectal (CCR). Inicialmente se observó una pérdida de metilación global en las células cancerosas de los pacientes con CCR en comparación con los controles sanos10. Al mismo tiempo, se comprobó que los promotores de los genes supresores tumorales aparecían hipermetilados, lo cual producía una menor expresión de dichos genes11. Estos hallazgos concluyeron con la asociación entre la hipermetilación de los genes supresores de tumores y el desarrollo de la enfermedad.
Sin embargo, en otras áreas de la medicina como las enfermedades neurológicas, todavía no se conoce bien la participación de la modificación en los patrones de metilación del ADN en el desarrollo de la enfermedad. En el caso concreto de la esclerosis múltiple (EM), recientemente se han encontrado modificaciones epigenéticas que podrían estar involucradas en la patogenia de la enfermedad, lo cual ha supuesto una nueva e interesante vía de investigación.
La EM se considera la primera causa de enfermedad neurológica grave que afecta a adulos jóvenes y de mediana edad. En España, los diferentes estudios de prevalencia realizados, muestran cifras en torno a 80 casos por 100.000 habitantes. Se trata de una enfermedad crónica que cursa con lesiones inflamatorias, desmielinizantes y neurodegenerativas en el sistema nervioso central (SNC). Su etiología es aún desconocida, aunque se presume un origen autoinmune y multifactorial, en la que se han descrito varios factores de susceptibilidad genética y ambiental. Teniendo en cuenta la complejidad de la enfermedad y la participación de diversos mecanismos etiológicos, tanto genéticos como ambientales, es lógico suponer que pueda existir una alteración en la regulación epigenética que participe en su desarrollo12,13.
Factores de riesgo de esclerosis múltiple y cambios epigenéticosEstudios epidemiológicos y de agregación familiar sugieren que hay una predisposición genética para padecer esta enfermedad. Sin embargo, hasta la fecha, el único locus asociado de forma consistente con la EM es el complejo mayor de histocompatibilidad (CMH). Esta predisposición se ha asociado al haplotipo «DR2» (HLA-DRB1*1501-DQA1*0102-DQB1*0602), el cual determina un riesgo relativo de 4 de presentar EM14. El desarrollo de nuevas tecnologías como las matrices o arrays de polimorfismos han permitido identificar nuevos genes candidatos localizados fuera de la región del CMH. De modo que la EM es una enfermedad poligénica en la que cada uno de los genes contribuye con un riesgo diferente (habitualmente bajo o moderado) a su desarrollo15.
Por lo tanto, sabemos que existe un factor genético de susceptibilidad de padecer EM, pero la presencia factores ambientales que provoquen cambios epigenéticos parecen imprescindibles para el desarrollo de la enfermedad16,17. A continuación, resumimos los 3 factores de riesgo ambiental descritos para la EM y los efectos que estos factores pueden ejercer sobre los diferentes mecanismos de regulación epigénetica, tanto en la EM como en el desarrollo de otras enfermedades18,19.
Tabaco y mecanismos epigenéticosEl tabaco supone uno de los factores ambientales que influye en el desarrollo de la EM, y así lo han demostrado distintos trabajos20,21. De hecho, se ha relacionado el hábito tabáquico con un aumento de la frecuencia de recaídas y del número de lesiones cerebrales activas en la RM craneal de los pacientes con esta enfermedad22.
Por otro lado, al analizar la sangre de adolescentes cuyas madres fumaron durante su embarazo, se ha observado que la exposición prenatal al tabaco se asocia con una mayor metilación del promotor del «brain derived neurotrophic factor» (BDNF), el cual, promueve la diferenciación y crecimiento de nuevas células neuronales23. Así mismo, en otro estudio llevado a cabo por Kjersti Aagaard et al., analizaron el patrón de metilación del ADN, mediante técnicas de PCR, en 2 grupos de sujetos fumadores y no fumadores. Encontraron alteraciones en la metilación del ADN en 25 genes de los no fumadores y en 438 genes de los fumadores24. También en el campo de la oncología se han encontrado cambios epigenéticos asociados al tabaco. En un estudio llevado a cabo en pacientes afectos de cáncer de pulmón se encontró una hipermetilación de los genes supresores de tumores CDKN2A, DAPK y MGMT de los pacientes fumadores25. Y, en un estudio realizado sobre el cáncer de cuello uterino, en mujeres de entre 15-19 años fumadoras, se encontró también una hipermetilación del gen CDKN2A en las células epiteliales cervicales de las fumadoras26.
Vitamina D y mecanismos epigenéticosEl déficit de vitamina D es uno de los factores de riesgo destacados en el desarrollo de la EM27–29. Esto se debe a que la vitamina D es un potente regulador de la respuesta inflamatoria e inmunomoduladora del cuerpo, actuando tanto sobre la inmunidad adaptativa como sobre la inmunidad innata30.
Aunque el mecanismo mediante el cual la vitamina D produce esos cambios es aún incierto, un estudio realizado por Joshi et al. sugiere que podría deberse a modificaciones epigenéticas. En dicho estudio se analizan los efectos de la 1,25(OH)2D3 (forma activa de la vitamina D producida en la piel tras la exposición a luz ultravioleta) en la producción humana de IL-17A mediante células T CD4+. Lo que observaron fue que la 1,25(OH)2D3 inhibe directamente el locus del IL-17 responsable de la transcripción de las citoquinas proinflamatorias mediante una modificación de la histona deacetilasa 2 (HDAC2) en la región promotora del IL17A31. Otros estudios ya habían mostrado previamente que la vitamina D es capaz de provocar cambios epigenéticos, como en el caso del cáncer de colon, donde se ha observado que la 1,25(OH)2D3 es capaz de inducir la expresión del gen que codifica la desmetilasa lisin-específica (JMJD3)32.
Virus Epstein Barr (VEB) y mecanismos epigenéticosHasta la fecha, varios agentes infecciosos han sido serológica y patológicamente asociados con la EM, muestra de esto, es un trabajo publicado por Sundstrom et al., en el que analizan las evidencias para apoyar si existe una infección viral en el estadio presintomático de la EM. Sin embargo, únicamente el antígeno extraíble del EBV presentó una correlación patológica directa con la aparición de la EM33,34.
De esta manera, la infección por VEB se ha asociado con cambios epigenéticos en las células infectadas. Varios tipos de tumores se han visto relacionados con la infección del VEB debido a una hipermetilación del promotor del gen supresor de tumores35, como ocurre en el cáncer de nasofaringe y el linfoma de Hodking inducidos por VEB, donde se ha visto que la hipermetilación del promotor es provocada por una elevación de las enzimas DNMT1, DNMT 3a y DNMT 3b, llevada a cabo a través de la proteína viral LMP136. En la EM los cambios epigenéticos relacionados con el VEB se asocian también con la expresión de microARNs (miARN). La expresión del miARN-142-3p en los pacientes con EM, se ha vinculado con una mayor tolerancia inmune, mientras que la expresión de miARN-155 se asocia con una mayor diferenciación de células T e inflamación del SNC37.
Metilación del ADN en la esclerosis múltipleSe desconoce el mecanismo fisiopatológico exacto que media entre los factores de riesgo ambiental y la susceptibilidad a desarrollar EM38. Y es precisamente en esta intersección donde la metilación del ADN puede aportar nuevos datos.
Inflamación y metilación del ADNEn los últimos años, diferentes autores han relacionado el grado de metilación en genes específicos con la presencia de la EM. En este sentido, Kumagai et al. han encontrado que el promotor del gen de la enzima esfingosina-1-fosfato (SPH-1), la cual participa en la regulación negativa de la señalización inflamatoria, se encuentra hipometilado en los pacientes con EM en comparación con controles sanos. La metilación del promotor del gen de la SPH-1 conduce a una disminución en la expresión de esta enzima y consecuentemente, a una mayor actividad de la inflamación mediada por los linfocitos39.
Por otro lado, Janson et al. han analizado los linfocitos T CD4+ de una serie de pacientes con EM recurrente remitente (EMRR), y lo que encuentran es que dichos pacientes presentan una desmetilación del gen FOXP3 que codifica para la proteína escurfina, cuya deficiencia está asociada a trastornos autoinmunes. Esta desmetilación del gen FOXP3 puede inhibir la diferenciación a células Th1 y Th2 y al mismo tiempo puede promover las células T reguladoras (Treg) y Th17. El equilibrio Th1/Th2 y Treg/Th17 influye en el estado de la enfermedad de manera que alteraciones en el mismo pueden conducir a la aparición de una nueva lesión o a su reparación, y es precisamente la metilación del ADN uno de los factores que regulan este equilibrio40,41.
También se ha encontrado una hipometilación del promotor del gen que codifica la IL-17A, citoquina proinflamatoria secretada exclusivamente por linfocitos T activados, que se ha relacionado con el desarrollo de enfermedades autoinmunes y que juega un papel central en la patogénesis de la EM42.
Desmielinización y metilación del ADNUn estudio llevado a cabo por Mastronardi et al. demuestra que durante el proceso de desmielinización de la sustancia blanca en pacientes con EM el promotor de la peptidil arginina deaminasa 2 (PAD-2) se encuentra desmetilado y, por lo tanto, la PAD-2 se sobreexpresa en el cerebro. Esta enzima provoca que la proteína básica de la mielina «myelin basic protein» (MBP) sea menos estable como consecuencia de la conversión enzimática de la arginina en citrulina. Esta citrulinización origina que la MBP se comporte como antígeno para los linfocitos T. En su análisis la metilación del promotor del PAD-2 de la sustancia blanca de pacientes con EM se halla reducida en un 25% respecto a los controles sanos. Además este cambio se encuentra solo en pacientes con EM y no en pacientes con otras enfermedades neurológicas como la enfermedad de Alzheimer, la enfermedad de Parkinson o la enfermedad de Huntington43.
Neurodegeneración y metilación del ADNHasta la fecha no disponemos de estudios que hayan analizado específicamente la participación de los mecanismos epigenéticos en el proceso de neurodegeneración de los pacientes con EM. Sin embargo, sí se han llevado a cabo análisis sobre los cambios en la metilación del ADN durante la muerte neuronal. Castaños et al. han analizado células de pacientes con esclerosis lateral amiotrófica (ELA) y han comprobado que la sobreexpresión de la enzima DNMT 3a induce degeneración y muerte celular, mientras que su inhibición las protege, y es precisamente la metilación del ADN la que regula la expresión de la DNMT3a44. Estos resultados nos orientan a pensar que dicho mecanismo podría estar implicado en el proceso de neurodegeneración que tiene lugar en los pacientes con EM.
ConclusionesLa EM supone una enfermedad neurológica con importante repercusión sanitaria, social y familiar, y a pesar de los importantes avances que se han producido estos últimos años, por el momento, desconocemos el mecanismo etiopatogénico exacto que causa la enfermedad y carecemos de un tratamiento curativo definitivo.
Los cambios epigenéticos, como la metilación del ADN, representan un mecanismo mediante el cual los factores ambientales pueden influir en la expresión génica individual. Resulta un campo de investigación de gran interés en el ámbito de la medicina, sobre todo en el estudio de aquellas enfermedades en las que se han detectado factores de riesgo ambiental que participan en su presentación y desarrollo, como ocurre en la EM.
Aunque el número de estudios realizados hasta la fecha en los pacientes con EM es escaso, sus resultados invitan a seguir profundizando en esta área. Los datos de los que disponemos en la actualidad señalan la existencia de una relación entre la regulación de la metilación del ADN en genes candidatos que son clave en el desarrollo de la EM y los procesos autoinmunes45,46. Si bien los resultados apuntan en esta línea, es necesario realizar estudios de cohortes con mayor número de pacientes y controles47,48.
El conocimiento de las modificaciones epigenéticas involucradas en la patogenia de la EM nos ayudará a clarificar los mecanismos de producción de la enfermedad y de esta forma nos abrirá el camino para la identificación de potenciales biomarcadores, así como para la búsqueda de nuevas dianas terapéuticas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Este trabajo no ha sido presentado en la reunión anual de la SEN, ni en otras reuniones o congresos. Para la realización de este trabajo no hemos recibido financiación de organismos públicos ni entidades privadas.

