




INTRODUCCION
Las inmunodeficiencias primarias (IDP) son un grupo de más de 100 enfermedades hereditarias, la mayoría de ellas monogénicas, que predisponen a sufrir infecciones recurrentes, alergias, procesos autoinmunes y cáncer1. Más del 60 % de estas enfermedades son procesos en los que predomina una deficiente formación de anticuerpos. Resulta paradójica la posibilidad de la presencia de una IDP de anticuerpos coexistiendo con una enfermedad autoinmune2. Sin embargo, nosotros describimos recientemente que un 17 % de pacientes con inmunodeficiencia variable común (IDVC) tuvieron como primera manifestación clínica un proceso autoinmune mediado por autoanticuerpos, sin haber tenido clínica infecciosa previa3. Tal hallazgo nos llevó a la realización del presente estudio con la finalidad de establecer la prevalencia de procesos autoinmunes en pacientes con diferentes IDP de anticuerpos. En relación con estudios previos no sólo incluimos a pacientes con IDVC o deficiencia selectiva de IgA4,5 sino también pacientes con deficiencias de subclases de IgG o con deficiencia selectiva de formación de anticuerpos. Tal información puede resultar útil tanto para la identificación de procesos autoinmunes en pacientes con IDP de anticuerpos, como para la posible detección de tales inmunodeficiencias en quienes cursan con enfermedad autoinmune.
MATERIAL Y MÉTODOS
Realizamos un estudio retrospectivo descriptivo de la prevalencia de datos clínicos y de laboratorio relacionados con autoinmunidad en un grupo de 152 pacientes adultos con IDP de anticuerpos. El diagnóstico de IDP de anticuerpos se realizó en la Unidad de Inmunología Clínica del hospital tras la evaluación clínica y realización de pruebas complementarias. Los pacientes procedían de distintos servicios clínicos del hospital. La distribución de las distintas inmunodeficiencias puede verse en la tabla I. Los diagnósticos de IDVC, deficiencia selectiva de IgA y agammaglobulinemia ligada al sexo se establecieron según los criterios del Grupo Panamericano de Inmunodeficiencias (PAGID) y de la Sociedad Europea de Inmunodeficiencias (ESID)6.

Consideramos deficiencia de subclases de IgG a la presencia de cifras disminuidas (al menos 2 DS por debajo del nivel normal) de alguna de las subclases de IgG (IgG1, IgG2 o IgG3), en presencia de niveles normales de IgG total, de IgA y de IgM y previa exclusión de causas conocidas de hipogammaglobulinemia, y en el caso de la subclase IgG4, el nivel indetectable como diagnóstico. El criterio diagnóstico de deficiencia específica de formación de anticuerpos fue la historia típica de defecto de la inmunidad humoral en asociación con incapacidad para la producción de anticuerpos anti-antígeno polisacárido (polisacárido de neumococo) o anti-antígeno proteico (toxoide tetánico) tras inmunización con el antígeno correspondiente, pese a tener niveles normales de IgG, IgA, IgM y de subclases de IgG.
La determinación de los niveles de inmunoglobulinas y subclases de IgG se realizó mediante técnica de nefelometría. La cuantificación del nivel de anticuerpos específicos se realizó mediante técnica de ELISA. Se consideró una respuesta normal de formación de anticuerpos cuando el nivel de anticuerpos detectado en el estudio basal aumentó un mínimo de tres veces 21 días tras la inmunización correspondiente. El diagnóstico de los distintos procesos autoinmunes se realizó también en la consulta de Inmunología Clínica con la excepción de aquellos casos que tenían ya diagnosticada la enfermedad autoinmune.
Los pacientes fueron clasificados como portadores de un proceso autoinmune según criterios vigentes para cada una de las entidades, entre ellos: criterios revisados de Sapporo para la clasificación del síndrome antifosfolípido7; criterios de la Academia Americana de Reumatología para la clasificación de lupus eritematoso sistémico, enfermedad lupus like, artritis reumatoide, artritis reumatoide juvenil, esclerodermia y vasculitis sistémica8-10.
La detección de autoanticuerpos incluyó las siguientes técnicas: Inmunofluorescencia indirecta (anticuerpos antinucleares, anti-músculo liso, anti-mitocondriales, anti-células parietales gástricas, anti-citoplasma de neutrófilos, anti-reticulina, anti-endomisio); ELISA [anticuerpos anti-cardiolipina, anti-antígenos nucleares extraíbles, anti-ADN, anti-tiroideos, anti-transglutaminasa, anti-gliadina (IgA o IgG), anti-insulina, anti-glutamato descarboxilasa (anti-GAD-65), anti-tirosina fosfatasa (anti-IA2)].
RESULTADOS
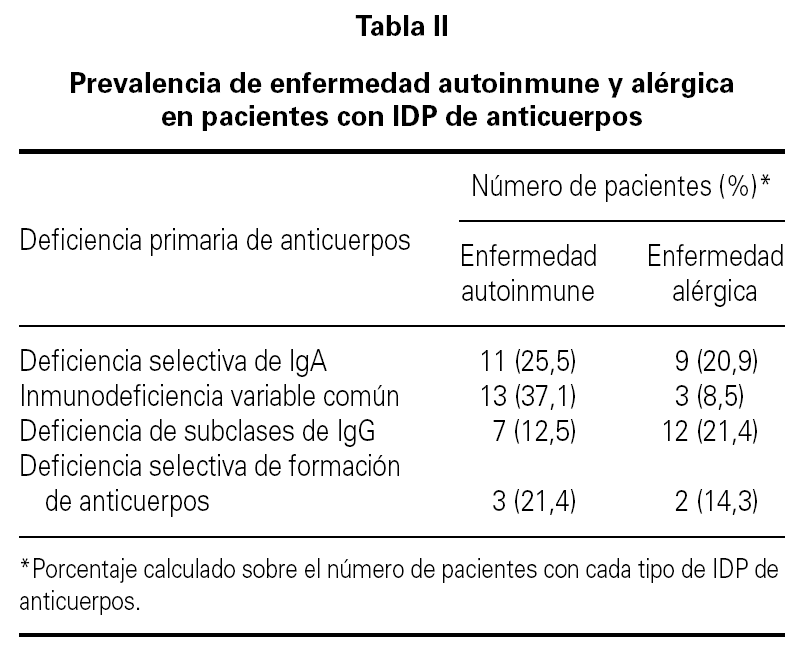
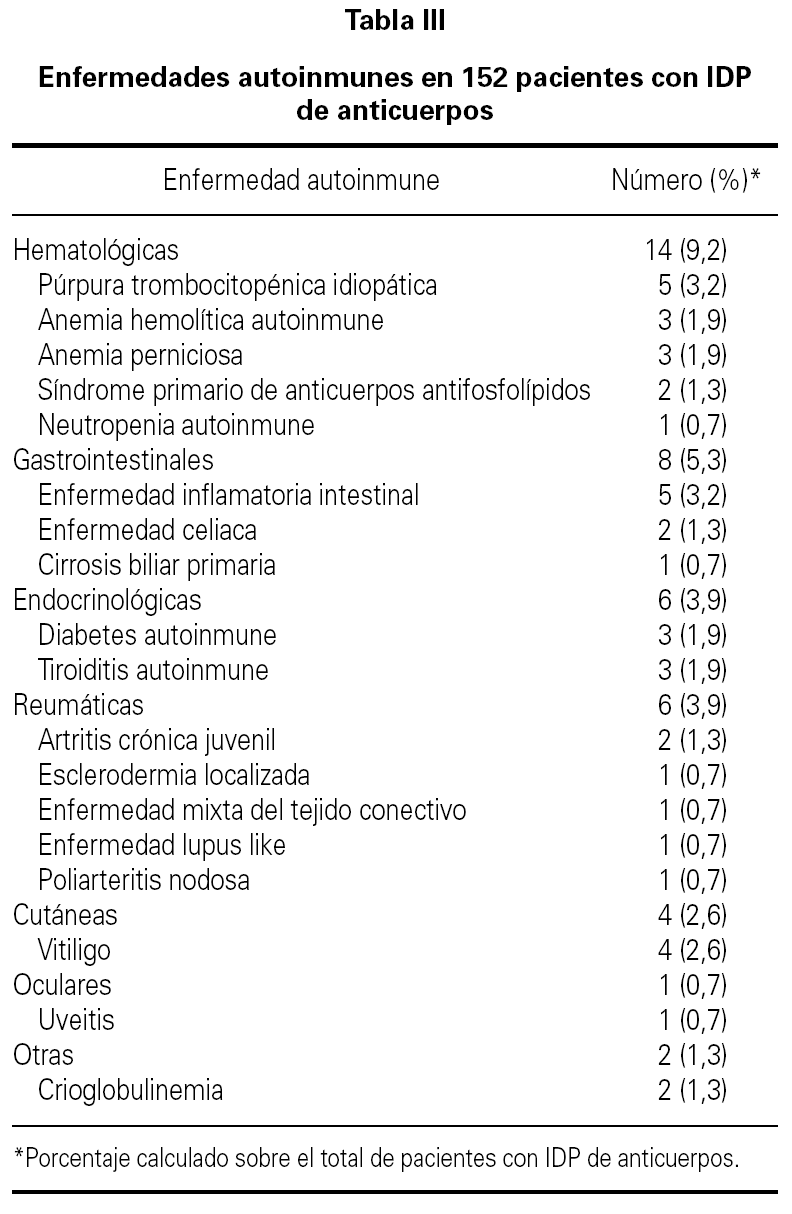
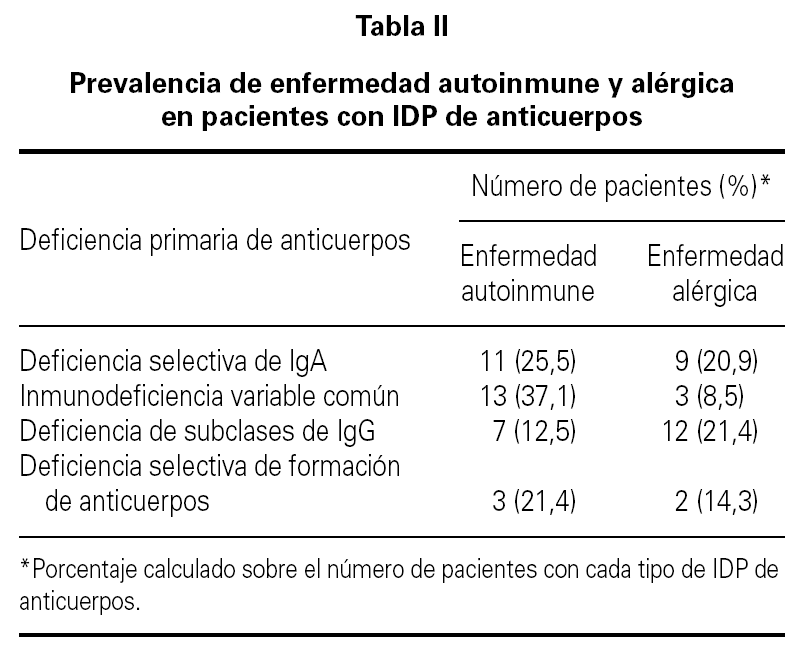
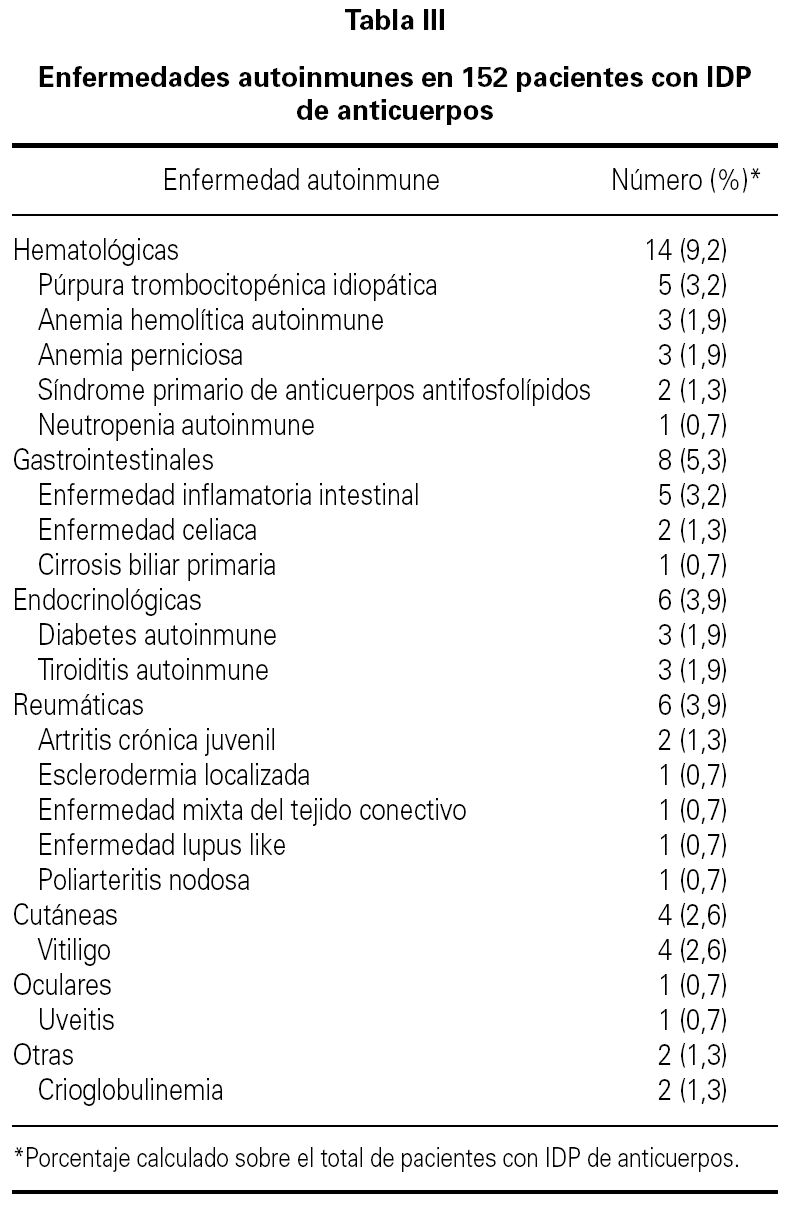
La media de edad de los pacientes fue de 40 años (intervalo 17-75 años) de los que 64 eran hombres (42 %) y 88 mujeres (58 %). El período medio de observación calculado desde el momento del diagnóstico de la IDP de anticuerpos hasta el momento de realización del estudio fue de 77 meses (intervalo 6-240 meses). 35 pacientes (23 %) tuvieron un proceso autoinmune a lo largo del curso evolutivo de la IDP de anticuerpos. En 13 de estos 35 enfermos (37 %) que tuvieron una enfermedad autoinmune, el diagnóstico de deficiencia de anticuerpos no era conocido en el momento del diagnóstico de la enfermedad autoinmune. La distribución de la prevalencia de enfermedad autoinmune en los cuatro grupos más frecuentes de IDP de anticuerpos se ve en la tabla II. Uno de los cuatro pacientes con agammaglobulinemia ligada al sexo tuvo un diagnóstico de artritis reumatoide juvenil. En otro paciente con agammaglobulinemia se desarrolló una enfermedad neurodegenerativa de etiología desconocida y falleció como consecuencia de la misma. La presencia de enfermedad autoinmune fue más frecuente en los pacientes con IDVC, aunque sin diferencia significativa en comparación con la observada en los que tenían deficiencia selectiva de IgA. Los pacientes con IDVC tuvieron una frecuencia significativamente mayor de proceso autoinmune que los afectos de déficit de subclases de IgG (p = 0,007). Seis de los 7 pacientes con deficiencia de subclases de IgG y enfermedad autoinmune asociada tenían deficiencia de la IgG4. En la tabla III se presentan las distintas enfermedades autoinmunes diagnosticadas. El grupo de enfermedad autoinmune más frecuente fue el de las citopenias autoinmunes seguido del de las enfermedades gastrointestinales, endocrinológicas y reumáticas. En 16 pacientes (10,5 %) se detectaron autoanticuerpos no asociados a datos clínicos de un proceso autoinmune concreto. Las especificidades de los anticuerpos se muestran en la tabla IV.

Otras enfermedades de base inmunológica prevalentes en los pacientes incluyeron: reacción de hipersensibilidad clase I en 26 (17 %) (asma, urticaria, angioedema); hiperplasia linfoide de tejidos en 8 (5,3 %), 7 de ellos en pacientes con IDVC y el otro con deficiencia selectiva de IgA. Asociaciones con procesos de posible base inmunológica fueron el liquen recurrente (2 pacientes con IDVC) o la psoriasis (2 pacientes con deficiencia selectiva de IgA). Se documentó un síndrome linfoproliferativo en un paciente con IDVC.
DISCUSION
La prevalencia de enfermedad autoinmune detectada en los pacientes con deficiencia de IgA y de IDVC es coherente con la descrita en trabajos previos2,4-5. Resulta interesante que en los pacientes con deficiencia de subclases de IgG también puede asociarse una elevada prevalencia de procesos autoinmunes, principalmente aquellos con deficiencia de IgG4, una deficiencia que ha sido cuestionada en cuanto a su significación patológica.
El diagnóstico de una posible enfermedad autoinmune debe sospecharse precozmente en pacientes que tienen una IDP de anticuerpos. Sin embargo, no contamos con marcadores para la identificación del riesgo de desarrollo de autoinmunidad en enfermos con IDP de anticuerpos. En pacientes con IDVC se ha sugerido que cambios en distintas subpoblaciones funcionales de células T reguladoras (CD4 + CD25 +)11, T activadas (CD4 + CD38 + DR + , CD8 + CD38 +)12 o en distintas subpoblaciones de linfocitos B definidas inmunofenotípicamente13, se asocian con la presencia de autoinmunidad. Controles periódicos son necesarios para detectar a los pacientes con IDP de anticuerpos que desarrollen un proceso autoinmune.
Por otro lado, tal como describimos en un estudio previo3 y confirmamos más ampliamente en el presente, las enfermedades autoinmunes pueden ser la primera manifestación de una IDP de anticuerpos. El mensaje en este sentido es la necesidad de controlar el nivel de inmunoglobulinas en el momento del diagnóstico de un proceso autoinmune, especialmente en aquellos casos en los que el enfermo va a ser sometido a tratamiento inmunosupresor. Estudios inmunológicos complementarios serán necesarios en aquellos pacientes con niveles normales de inmunoglobulinas en quienes se sospeche de la existencia de una inmunodeficiencia.
AGRADECIMIENTOS
Los autores agradecen a los Drs. Carmen Rodríguez-Perez, Silvia Sánchez-Ramón, Elena Seoane, Jose Ruiz-Tíscar, Jesús Bermejo, Dariela Micheloud y Juana Gil por su contribución al atender pacientes en la Consulta de Inmunología Clínica.
