


INTRODUCCION
La inmunodeficiencia común variable (1-4) es una inmunodeficiencia primaria (IDP) caracterizada por una deficiente producción de anticuerpos. Se trata de una hipogammaglobulinemia que afecta varones y mujeres y que puede presentarse a cualquier edad, siendo más frecuente entre la segunda y la tercera décadas de la vida. Aproximadamente afecta a 1 de cada 25.000 a 100.000 individuos de raza caucásica (según registros). No se conoce todavía el defecto o defectos genéticos, aunque se han descrito diversas alteraciones tanto en la respuesta humoral como en la función de las céulas T y síntesis de citocinas (5-8). La expresión clínica es variable, siendo la manifestación más frecuente las infecciones bacterianas de repetición, en especial de vías respiratorias (sinusitis, otitis, bronquitis y neumonías) por Haemophilus influenzae y otros gérmenes encapsulados y también del aparato gastrointestinal (por Giardia lamblia, Salmonella, etc.). Las septicemias y las infecciones del sistema nervioso central son raras, así como las infecciones vírales. Por otra parte las enfermedades autoinmunes y los procesos alérgicos se observan con una frecuencia superior a la población normal. También hay una mayor incidencia de procesos tumorales, en especial síndromes linfoproliferativos.
El diagnóstico de la inmunodeficiencia común variable (IDCV) (4) se basa en una cifra baja de inmunoglobulinas, con un defecto en la producción de anticuerpos (tanto frente a vacunas como a infecciones demostradas), una clínica compatible y la exclusión de otras enfermedades.
El diagnóstico diferencial debe establecerse sobre todo con otras IDP: hipogammaglobulinemia transitoria de la infancia, agammaglobulinemia ligada al cromosoma X (ALX), déficit de subclases de IgG, déficit de IgA (DIgA), hiper-IgM (HIM-X).
Una entidad propia de la edad pediátrica es la hipogamaglobulinemia transitoria de la infancia (9): se trata de pacientes menores de 3 años en los que los valores de las inmunoglobulinas séricas son bajos, con o sin clínica infecciosa y con células B presentes. En estos casos se aprecia una normalización progresiva de las inmunoglobulinas con desaparición espontánea de toda sintomatología en el trascurso de unos meses.
El diagnóstico de ALX se plantea sólo en los pacientes de sexo masculino y se basa en la ausencia de linfocitos B y una cifra baja de todas la inmunoglobulinas, debiendo prestarse atención especial a la cifra de IgM, puesto que en la IDCV suele estar más conservada. Además la sintomatología habitualmente es más grave y existen antecedentes familiares hasta en 2/3 de los casos. Por otra parte, actualmente el diagnóstico molecular con la detección de las mutaciones en el gen de la tirosin-cinasa es posible esos antecedentes existan hasta en el 80-90 % de los cases de ALX (10).
El déficit de subclases de IgG se caracteriza por una cifra significativamente baja de una o más subclases de IgG, en especial IgG1 e IgG2, mientras la IgG total es normal, baja o incluso alta, con defecto en la síntesis de anticuerpos. La respuesta de las células T es normal si bien pueden presentar linfopenia, especialmente de CD4 y anomalías en la síntesis de citocinas (11). El déficit de subclases de IgG, especialmente de IgG2 puede asociarse con DIgA y clínicamente suele expresarse con infecciones bacterianas de repetición y procesos autoinmunes.
El diagnóstico del DIgA se establece cuando la cifra de IgA es inferior a 5 mg/dl (11). Se trata de la IDP más frecuente. Generalmente es asintomática pero existe un incremento de las infecciones sinopulmonares y digestivas, procesos alérgicos y autoinmunes. El resto de las inmunoglobulinas son normales o altas, la respuesta celular es normal así como la síntesis de anticuerpos.
El HIM-X se diferencia por la cifra alta de IgM, con ausencia de IgG e IgA y células B presentes. En los casos de HIM ligados al cromosoma X también es posible el diagnóstico molecular de los defectos de expresión del CD40L (CD154)y posterior estudio genético (12).
Finalmente también deberán descartarse otros procesos que cursan con infecciones respiratorias de repetición, como la fibrosis quística y el síndrome del cilio inmóvil.
El tratamiento de la IDCV, así como del defecto de subclases de IgG con defecto en la producción de anticuerpos, asociado o no al DIgA, es la administración de anticuerpos en forma de gammaglobulina intravenosa (GGIV) cada 2-4 semanas, siendo la dosis más usual entre 200-400 mg/kg de peso (13). Con ello se consigue prevenir las complicaciones a largo plazo de las infecciones y mejora la calidad de vida de los pacientes. Es por esta razón que el diagnóstico precoz es esencial.
MATERIAL Y MÉTODOS
Se realizó un estudio retrospectivo de los 16 casos diagnosticados de IDCV en nuestro Hospital Infantil en los últimos 20 años (1980-2000).
De las historias clínicas se recogieron: edad al diagnóstico y al inicio del tratamiento con GGIV, tiempo de seguimiento, infecciones (previas al tratamiento y posteriores al mismo), existencia o no de bronquiectasias, cultivos positivos (antes y después del inicio del tratamiento con GGIV), presencia de procesos autoinmunes, alérgicos, tumorales u otros (antes y después del diagnóstico), antecedentes familiares, niveles de inmunoglobulinas (IgG, IgA, IgM, subclases de IgG) al diagnóstico y a los 6 meses o más del inicio del tratamiento, fenotipo linfocitario (linfocitos T, CD4, CD8 y linfocitos B)y autoanticuerpos (ANA anti-DNA).
Los valores de inmunoglobulinas se midieron por nefelometría; las subclases de IgG por ELISA con anticuerpos monoclonales (AcMo); el fenotipo de los linfocitos en sangre periférica por citometría de flujo (FACScan) con AcMo CD3 + , CD4 + , CD8 + y CD19 +; los ANA por inmunofluorescencia indirecta y los anticuerpos anti-DNA por radioinmunoensayo.
RESULTADOS Y DISCUSION
Se observa un predominio del sexo masculino (11/16, 68,75 %). La edad al diagnóstico osciló entre 7 meses y 15 años. La edad media al diagnóstico fue de 8,5 años.
Respecto al paciente cuyo diagnóstico se produjo a los 7 meses debe decirse que además de la sospecha clínica (celulitis de repetición con cultivos positivos a H. influenzae) se añadieron los antecedentes familiares (hermano afecto también de IDCV) que favorecieron su diagnóstico precoz. Aunque a esa edad la hipogammaglobulinemia transitoria de la infancia es el primer diagnóstico, el seguimiento posterior ha confirmado la IDCV.
El tiempo transcurrido entre el inicio de sintomatología valorable y el inicio del tratamiento osciló entre 2 meses y 112 meses, siendo la media de 45 meses. En la mayoría de casos el diagnóstico se realizó peco tiempo antes del inicio del tratamiento, sólo en 2 casos se retrasó por negativa familiar. Así pues, en general la sospecha de IDP y por consiguiente su estudio se producen con bastantes meses de demora.
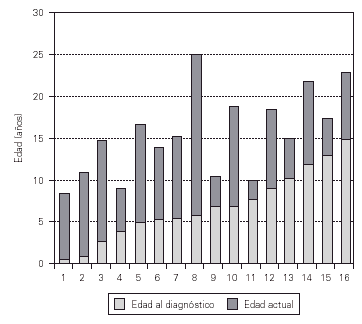
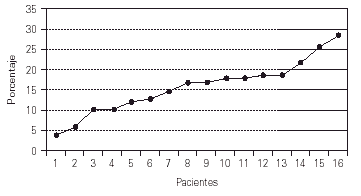
El tiempo de seguimiento de los pacientes oscila desde 2 a 19 años (media 8 años) (fig. 1).
Figura 1.--Tiempo de seguimiento de los pacientes.
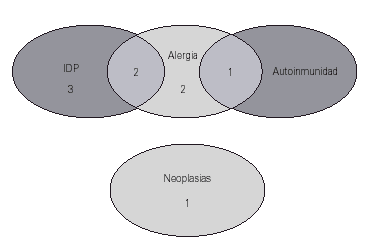
Entre los antecedentes familiares de primer grado destaca que en 5 casos (31 %) hubo otras inmunodeficiencias primarias en uno o varios familiares: déficit de IgA y otras IDVC. Este dato apoya la relación que diversos investigadores (10, 11) proponen como dos formas de distinta gravedad de un defecto primario similar. En 5 de los 16 pacientes había antecedentes familiares de alergia, en un caso de enfermedad autoinmune y en otro caso neoplasia. En la figura 2 se recogen las distintas asociaciones de estos antecedentes en los pacientes: en dos de ellos los antecedentes fueron de otras IDP y alergia, y en un caso de alergia y autoinmunidad.
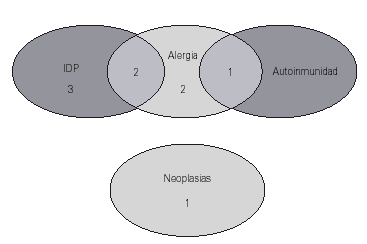
Figura 2.--Antecedentes familiares.
Entre las manifestaciones clínicas antes del diagnóstico destaca la presencia de infecciones, siendo las más frecuentes las respiratorias de repetición (81 %)y las gastrointestinales (57 %), asociándose ambas en muchos casos. Este predominio de la incidencia de infecciones respiratorias ya se anotó en el momento del diagnóstico: 33 % infecciones respiratorias de vías bajas, 25 % infecciones respiratorias de vías altas (sinusitis, otitis), 28 % infecciones gastrointestinales, 14 % infecciones cutáneas. No se detectó ningún caso de sepsis, osteomielitis, infecciones del SNC ni urinarias.
Entre los cultivos positivos el H. influenzae fue el germen más frecuente, seguido del Pneumococo, en vías respiratorias. Otros cultivos mostraron G. lamblia en procesos gastrointestinales.
Durante el seguimiento se han diagnosticado 5 casos de bronquiectasias (31 %), cuyos pacientes tenían una edad que oscilaba entre 3 y 11 años (media: 7 años).
En 7 de los pacientes (43,75 %) hubo manifestaciones clínicas de alergia: seis casos de asma, dos de eccema atópico, uno de urticaria, uno de alergia medicamentosa, uno de anafilaxia. Como se observa en algún paciente se asociaban diversas manifestaciones alérgicas, siendo la mayoría asmáticos. Uno de los pacientes afectos de asma presentaba un prick-test positivo a pneumoalergenos (gramíneas y olivo), en los pacientes restantes no consta si se realizó la prueba o bien fue negativa. Los 2 casos de eccema y la urticaria aguda evolucionaron favorablemente con tratamiento sintomático y la administración de GGIV. En el paciente afecto de asma y alergia a medicamentos se mantiene la sintomatología y requiere tratamiento inhalado además de la GGIV. El caso en el que hubo una reacción anafiláctica, ésta ocurrió durante la administración de GGIV en otro Centro y posteriormente no se ha repetido.
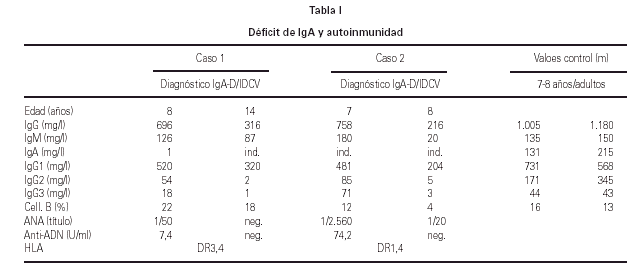
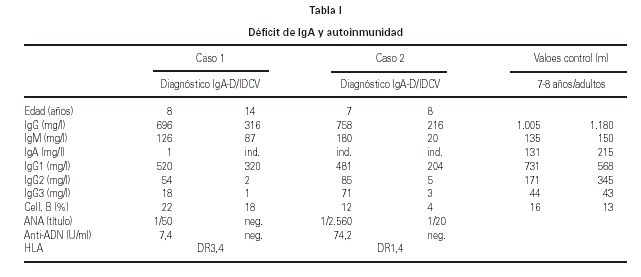
Se diagnosticaron 3 procesos autoinmunes (19 %) (16, 17) y en un caso se hallaron autoanticuerpos positivos séricos, sin que tuviese manifestaciones clínicas durante su seguimiento. De los casos diagnosticados hubo: un caso de hepatitis autoinmune y vitíligo, un caso de hepatitis autoinmune y otro de lupus eritematoso sistémico. Tanto estos tres pacientes como en el que se detectaron autoanticuerpos se trata de pacientes del sexo femenino. Todos ellos evolucionaron favorablemente con el tratamiento adecuado y actualmente sólo precisan tratamiento sustitutivo con GGIV sin necesitar inmunosupresores. Cabe destacar que en 2 de estas pacientes se diagnosticó el déficit de IgA 1 y 6 años respectivamente antes que la IDCV (15) (tabla I).
No hubo ningún caso de neoplasia ni tampoco falleció ninguno de los pacientes.
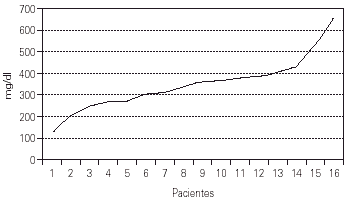
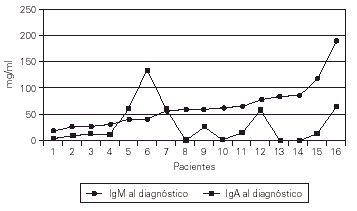
En los datos de laboratorio cabe destacar los valores de IgG al hacer el diagnóstico: entre 200 y 400 mg/dl, con cifras límite de 125 y 625 mg/dl, debido a la variabilidad propia de la edad en que se determinaron. Hay que añadir que en muchos casos se detectó también la mala calidad de los anticuerpos existentes, con escasa respuesta a vacunas administradas previamente (tétanos, polio, etc.) o antígenos naturales (fig. 3).

Figura 3.--Valores de la IgG al efectuar el diagnóstico.
Las cifras de IgA al hacer el diagnóstico eran indetectables en 5 casos (31 %)y muy bajas en otros 6. La IgM al diagnóstico era normal o ligeramente baja, a diferencia de la AXL y la HIM-X (fig. 4).
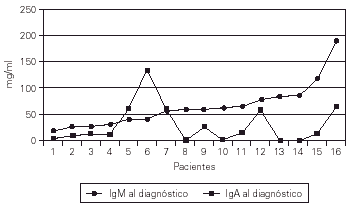
Figura 4.--Valores de la IgA e IgM al efectuar el diagnóstico.
En cuanto a la inmunidad celular, el porcentaje de linfocitos B (CD19) osciló entre 4 y 29 %, es decir, valores normales o bajos. Se ha descartado el diagnóstico de agammaglobulinemia ligada al X en los que presentan linfocitos B inferiores al 2 % (fig. 5).
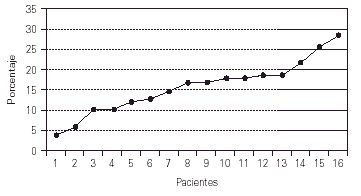
Figura 5.--CD19+ al efectuar el diagnóstico.
CONCLUSIONES
La manifestación clínica más frecuente en los casos diagnosticados de IDVC han sido las infecciones respiratorias de repetición, tanto de vías aéreas superiores como inferiores.
Destaca también la elevada incidencia de bronquiectasias (31 %), a pesar del diagnóstico a temprana edad y tratamiento adecuado.
Se encontró asimismo un aumento de procesos alérgicos y autoinmunes, que en algunos casos fue la primera manifestación clínica de la IDCV.
Por otra parte existe relación entre el déficit de IgA y la IDCV por dos vertientes distintas: en primer lugar por los antecedentes familiares (hay casos que comparten familiares con ambas IDP)y en segundo lugar porque se ha documentado el déficit de IgA aislado antes del diagnóstico de IDCV en 2 de los pacientes.
Por último resaltar la necesidad del diagnóstico precoz y tratamiento adecuado con gammaglobulinas IV para evitar las secuelas a largo plazo y mejorar la calidad de vida de los pacientes.
