



Non-alcoholic fatty liver disease (NAFLD) is considered to be a manifestation of liver metabolic damage and is related to insulin resistance and genetic susceptibility. Inflammation mediated by Kupffer cells (KCs) is of critical importance to the development of NAFLD. The primary role of KCs in NAFLD is considered to be the perturbation of the C-Jun N-terminal kinase (JNK) and nuclear factor-kappa B (NF-κB) pathways as a result of lipopolysaccharide (LPS) recognition by Toll-like receptor 4 (TLR4). Simultaneously, the activation of NF-κB, as mediated by oxidative and endoplasmic reticulum (ER) stress and free fatty acid (FFA) or free cholesterol (FC) crystal formation, heavily relies on NF-kB regulatory factors and TLR4. Additionally, the imbalance of certain pro-inflammatory cytokines and chemokines released by innate immunity is deemed to promote the steatosis of hepatocytes. In conclusion, this review indicates that the inflammatory and oxidative stress of KCs play a significant role in the development of NAFLD.
Non-alcoholic fatty liver disease (NAFLD), a type of metabolic liver damage that has affected Chinese citizens for years, is related to insulin resistance and genetic susceptibility.1 NAFLD includes nonalcoholic fatty liver (NAFL) and nonalcoholic steatohepatitis (NASH) and is related to liver cirrhosis and hepatocellular carcinoma (HCC) according to its histological classification.2 The emergence of steatohepatitis without hepatocellular injury is categorized as NAFL. Meanwhile, NASH is histologically further defined as the emergence of inflammation and steatohepatitis with injury or fibrosis of hepatocytes. The histological shift of NAFL-to-NASH is primarily known as the “double hit” theory.3 The ”first hit” is characterized by multiple metabolic syndromes and insulin resistance, which are caused by free fatty acid (FFA) and lipid accumulation in peripheral blood and hepatocytes. The “second hit” refers to a series of innate immune responses in leukocytes, such as Kupffer cells (KCs), that are caused by the stimulation of lipotoxins and lipopolysaccharide (LPS), ultimately leading to steatohepatitis, fibrosis and other irreversible liver pathologies.4,5
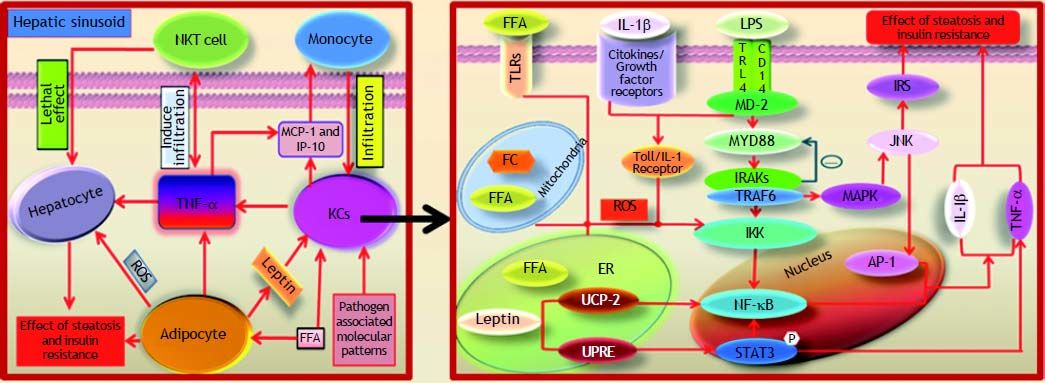
KCs are the resident macrophages in liver tissue that prevent harmful endotoxins present in the portal vein from entering into the circulation. Inflammation mediated by KCs is of critical importance in the development of NAFLD. Using chemicals to delete KCs has been demonstrated to alter the release of pro-inflammatory cytokines and to alleviate hepatocellular damage.6 Ono and colleagues have suggested that the “second hit” plays a key role in the NAFL-to-NASH transition.7 They have determined that the phagocytic dysfunction of KCs can accelerate inflammatory necrosis during hepatocyte fat accumulation and that the ED2+ KCs play a greater role in the pathological progression of NAFLD. The ED2+ KCs, which are also known as alternatively activated M2 KCs, show higher immunobiologic activity than ED1+ KCs (classical Ml KCs) with close correlation to steatohepatitis severity. Recent reports suggest that the regulation of the M1/M2 KCs balance in hepatocytes with steatohepatitis, which results in apoptotic effects of M2 KCs, reverts KCs toward their M1 KCs counterparts.8,9 However, little is known about the intracellular signaling pathways of KCs that are mediated by LPS, FFA and ER stress, for example, and how they are involved in NAFLD. This topic is the centerpiece of this review.
The Effect of KCS Induced by LPS on NAFLDEnterogenous bacterial components such as LPS play a key role in the pathogenesis of NAFLD, as previously reported.10–12 Studies suggest that the gut-liver axis is mainly induced by probiotics in the pathogenesis of NAFLD.13 Conversely, the increase in intestinal permeability as a result of a high-glucose and high-fat diet leads to the accumulation of enterogenous LPS, which irritates the innate hepatic immune response.10,11 The binding of LPS and the receptor complexes on the surface of KCs activates pro-inflammatory cytokines that then recruit T lymphocytes, B lymphocytes and other leukocytes.12 The aggregation of immune cells in liver tissue easily triggers steatohepatitis and inflammatory necrosis in hepatocytes, followed by NASH progression.14 LPS is eliminated by KCs in the final barrier preventing the spread of LPS from the portal vein to peripheral circulation.10–11 Using a mouse model of NAFLD, Imajo, et al. have demonstrated a hyper-reaction to a small dose of LPS that is mediated by the signal transducers and activators of transcription 3 (STAT3) pathway induced by the leptin pathway.15 The continuous activation of KCs by LPS leads to the up-regulation of downstream signaling molecules, such as tumor necrosis factor α (TNF-α), which aggravates steatohepatitis and inflammatory necrosis in hepatocytes.
CD14, a component of membrane receptor complexes, is essential for KCs to bind to LPS. There are two reported types of CD14: mCD14, which is anchored to the KC membrane via a glycosylphosphatidylinositol tail and sCD14, a serum-soluble form of the protein that lacks glycolipid tail found in mCD14.16–19 Ogawa and colleagues have recently reported that serum CD14 may be a potential marker for necrotic liver inflammation in NAFLD mice.20 Moreover, they also suggest that TNF-α is increased by the activation of NF-κB inhibition factor kinases (IKK) as a result of the binding of LPS and CD14 on the membrane of KCs. Tonan, et al. have demonstrated that CD14 expression correlates with KCs phagocytic function in vitro.21 The phagocytic deficiency prevents KCs from removing LPS, further promoting the generation of pro-inflammatory cytokines. Fukada, et al. have found that the LPS-mediated activation of KCs inhibits cell au-tophagy, which might augment the sensitivity of KCs to LPS in the model of NAFLD.22 Their report suggests that the metabolic disorder of lipid and LPS disrupts KCs in steatohepatitis; however, the mechanism by which KCs become dysfunctional following attack by LPS and lipids remains unclear.
The identification of pathogen associated toll-like receptors (TLRs) shows that they participate in the activation of C-Jun N-terminal kinase (JNK) and NF-kB.23 TLR4 is highly expressed in the NASH mouse model compared with other types of TLRs.23–24 TLR4 conducts the transmembrane signal as a component of the membrane receptor complexes that bind to LPS.23 Enterogenous endotoxins in NAFLD mice promote the progress of steatohepatitis via the myeloid differentiation molecular 88 (MYD88)-de-pendent TLR4 signaling pathway.25 The binding of LPS to TLR4 recruits MYD88 to bind interleukin receptor associative kinases (IRAKs) and tumor necrosis factor receptor associated factor 6 (TRAF-6).26,27 Pro-inflammatory cytokines, such as TNF-α, Interleukin-6 (IL-6) and IL10, are increased by the activation of JNK/MAPK and IKK. The complex also activates the MYD88-independent pathway, which is mediated by the combined action of the Toll/IL-1 receptor, which is induced by interferon in response to the IKK ligand binding. Furthermore, the activation of interferon-β (IFN-β) mediated by the MYD88-independent pathway, is involved in the inflammatory response of NAFLD.28 Thus, the activation of KCs generates pro-inflammatory cytokines such as TNF-α, IL-1β, IL-2, IL-6, IL-10 and IFN-γ, which promote the infiltration of neutrophils, natural killer cells (NKCs), natural killer T cells and T cells, among others.29,30 TNF-α, which is released by KCs, accelerates the process of NAFLD, triggering monocyte infiltration through the expression of interferon inducible protein 10 (IP-10) and macrophage chemotactic protein 1 (MCP-1).30 Ajamieh, et al. have reported that the expression of adhesion molecules, cytokines, chemokines and thromboxane B2 (TXB2) is suppressed by inhibiting the activation of NF-kB through the decreased expression of TLR4.31 X-box binding protein 1 (XBP-1), which is TLR4-dependent, is activated by the oxidative stress caused by the innate immune response of KCs and strongly promotes the NALF to NASH progression.24 The receptor complexes of LPS are essential in the initiation of the innate immune response. Prolonged inflammation, as mediated by LPS irritation, easily induces irreversible necrosis and fibrosis in hepatocytes (Figure 1).

Liver X receptor α (LXRα), a member of metabolic nuclear receptors, plays a significant role in lipid metabolism, especially in the regulation of cholesterol secretion and metabolism.32 Recently, studies have suggested that LXRα plays a protective role in NAFLD and bridges fat metabolism with inflammatory responses in the liver.33 LXRα exerts an anti-inflammatory effect through the NF-kB pathway in KCs. TLR4, IL-lβ and TNFα, which are activated by NF-kB signaling pathways, are suppressed by the activation of LXRa.34,35 The down-regulation of glutamate receptors interacting with protein3 (GRIP3), interferon regulatory factor3 (IRF3) and IRAK4 on KCs reduces hepatic injury in NAFLD mice.36 IL-lβ, cyclooxygenase 2 (COX-2), inducible nitric oxide synthase (iNOS), matrix metalloprotease 9 (MMP-9) and others are inhibited by ligand-activated LXRα.37 Xu, et al. consider LXRα/sterol regulatory element binding protein1-c (SREBP1-c) to be the most significant point in the protective mechanism of LXRα in NAFLD.38 SREBPl-c is up-regulated in the process of lipid synthesis and increases the expression of LXRα.39 SREBP1-c is highly expressed in both adipose and liver tissue and is known to be the key molecular signal in insulin resistance.40 SREBP-1c is highly expressed in hepatocytes with steatohepatitis to increase fatty acid synthesis and to inhibit β-oxidation by TNFα.41–43 The target genes of LXRα in the regulation of inflammation and lipid metabolism are unclear. However, high levels of oxygen oxysterol, the ligand of LXRα in the peripheral circulation, may be important in insulin resistance.
The Influence of FFA and FC Assembled in KCsKCs are not increased in liver tissue bordering steatohepatitis; however, fat-laden KCs or KCs with a significant accumulation of intracellular toxic lipids were found in a mouse model of NAFLD.44 TNF-α is highly expressed by the FFA-activated NF-κB pathway in fat-laden KCs.45 The immunological competence of KCs is disrupted by the excess lipid accumulation in liver tissue via the following mechanisms. Leukocytes in the liver sinusoids reinforce the inflammatory response of KCs in microvascular vessels i.e., the liver innate immune response.46 Lipids regulate inflammation and insulin resistance by their interaction with receptors, such as TLR4 and LXRα, on either the inside or outside of KCs.47 Excessive lipid accumulation in the cellular plasma membrane changes the structure of the lipid rafts and influences the aggregation and function of cell membrane receptors. This accumulation also affects the function of cholesterol-free plasma membranes, such as mitochondria, resulting in oxidative and endoplasmic reticulum (ER) stress.48 Abnormal lipid accumulation interferes with the identification of KCs for hepatocytes with the fat degeneration, which may be associated with the dysfunctional phagocytosis of KCs.49
The binding of extracellular FFA by TLRs on KCs activates the JNK and NF-κB pathways. Adhesion molecules and MCP-l are up-regulated by activated NF-κB, which recruits CD11b+ macrophages and promotes lipid synthesis, thereby elevating the transcription of activating protein 1 (AP-1) and proinflammatory cytokine.45,50,51 Excessive FFA in KCs impairs β-oxidation and other functions of mitochondria.52 Alternatively, oxidative and ER stress are provoked by the JNK and NF-κB pathways, inducing pro-inflammatory signals and insulin resistance.51 Fat-laden KCs evolve into M2 KCs, which enrich lymphocytes by LPS stimulation.44 In fact, this phenotype of KCs is important during the early stages of innate immunity in steatohepatitis (Figure 1).
The downstream molecular signals of active NF-κB are perturbed as a direct or indirect effect of FC crystals on IKK.14 Accumulating FC has been proven to be the lipotoxin in fatty liver that is the main cause of insulin resistance.53–55 Interestingly, Bieghs, et al. have insisted that the distribution of cholesterol in vivo is under the control of CD36 and macrophage scavenger receptors on KCs. These receptors are closely related to the lysosome pathway.56 Goudriaan, et al. have discovered that liver insulin resistance may be induced by CD36 deficiency but that insulin sensitivity increases in the muscle of CD36-/- mice.57 However, this mechanism could stimulate the inflammatory response of NAFLD, indicating that triglycerides may play an important role in disease progression. However, current studies suggest that excessive triglycerides protect the liver from NAFLD via the high expression of LXRa, rather than injuring hepatocytes through inflammation or fibrosis.14,58
The Function of Oxidative Stress in Kcs on NAFLDThe excessive aggregation of FFA and FC in KCs leads to steatohepatitis and the formation of fatladen KCs.44 Oxidative stress is induced by insufficient FFA β-oxidation or dysfunction resulting in the lipotoxicity of the mitochondria, which triggers the activation of the NF-κB/JNK pathway, high mobility group box 1 (HMGB-1)/TLRs, cytokines and chemokines.14,44,56,59 Our group has demonstrated that LPS induces the relocation and release of HMGB1 by activating the NF-κB signaling pathway.60 Further evidence suggests that the imbalance of antioxidants and peroxide in fat-laden KCs leads to membrane damage, DNA or protein synthesis in hepatocytes and cytokine cascade dysregulation eventually prompting the progression of hepatic fibrosis.61–63 Uncoupling protein 2 (UCP-2), which is largely inhibited by KCs, is anchored in the mitochondrial inner membrane of hepatocytes and is induced by FFA via PPAR-α.64–66 In an ER laden with excess FFA, ER stress is induced by an insufficiency or dysfunction in the unfolded protein response element (UPRE), which in turn activates the JNK/ NF-κB/(C/EBP) pathway. Insulin resistance is initiated by the activation of insulin receptor substrate 1(IRS-1) and IRS-2 via the JNK pathway.14,67 Bc1-2, an apoptotic inhibitory factor, is inhibited by C/EBP, which increases the viability of the proapoptotic protein Bim.68 Apoptosis and fibrosis in hepatocytes are hallmark pathological features of NAFLD. However, the mechanism by which FFA accumulation in the ER of KCs induces ER stress is unclear, and there is insufficient evidence for a relationship of metabolic syndrome with NAFLD (Figure 1).
The Other Functions of KCs on NAFLDDuring steatohepatitis, KCs highly express membrane receptors and generate excessive levels of cytokines, chemokines, arachidonic acid, proteolytic enzymes, peroxide and nitric oxide.69 The recruitment of lymphocytes, leukocytes and macrophages during steatosis increases cytotoxicity and the innate immune response, thereby promoting NAFLD.14 IL-1 and IL-18 are produced by KCs recruiting T lymphocytes and natural killer cells to the liver, while INF-γ kills the steatotic hepatocytes and regulates active T cells responses.70 However, the subset of cells and the specific pathological effect of T cells on the progression of NAFLD remain unknown. Tang, et al. have demonstrated that the number of M2 KCs increases in mice fed a high-fat diet, secreting TNF-α to activate NKCs that hepatocytes.71 Neutrophils have been reported to cause hepatic necroinflammation in the NAFLD mouse model, but this mechanism remains unclear.72
In conclusion, the mechanisms involved in KCs-mediated NAFLD development are as follows: KCs are activated by the binding of LPS or FFA to TLRs and release cytokines and chemokines via the NF-κB signaling pathway. The activation of the NF-κB signaling pathway is also directly induced by oxidative or ER stress when LPS or FFA binds to TLRs. Several pro-inflammatory cytokines, such as TNF-α, IL-1β, IL-6, C-C chemokine receptors 2 (CCR-2), macrophage inflammatory protein 1 (MIP-1), COX-2, MCP-1 and intercellular adhesion molecule/vascular adhesion molecule (ICAM/VCAM), are produced by the activated NF-κB pathway. NKCs, natural killer T are assembled by the aforementioned pro-inflammatory cytokines and infiltrate the live tissue, resulting in an imbalance of downstream signaling molecules. These pro-inflammatory cytokines, particularly TNF-α, are released by KCs and lead to cytolysis, dead hepatocytes and inflammatory necrosis, eventually resulting in insulin resistance and liver fibrosis. Furthermore, TNF-α antibodies that target KCs are proposed to be an effective treatment for severe NAFLD patients in the near future.30 Meanwhile, pentoxifylline, which suppresses TNF-a synthesis, attenuates free radical mediated excessive lipid oxidation in NASH patients.73 Despite small scale experiments in humans that have indicated that pentoxifylline may be a valid NAFLD therapy, employing this drug in clinical practice has yet reveal the “smoking gun”.74,75
Abbreviations- •
AP-1: activating protein 1.
- •
CCR-2: C-C chemokine receptors 2.
- •
COX-2: cyclooxygenase 2.
- •
ER: endoplasmic reticulum.
- •
FC: free cholesterol.
- •
FFA: free fatty acid.
- •
GRIP3: glutamate receptors interacting with protein3.
- •
HMGB-1: high mobility group box1.
- •
HCC: hepatocellular carcinoma.
- •
ICAM/VCAM: intercellular adhesion molecule/ vascular adhesion molecule.
- •
IFN: interferon.
- •
IKK: NF-κB inhibition factors kinase.
- •
IL: interleukin.
- •
IP-10: inducible protein.
- •
IRAKs: interleukin receptor associative kinases.
- •
IRF3: interferon regulatory factor3.
- •
IRS-1: insulin receptor substrate 1.
- •
JNK: C-Jun N-terminal kinase.
- •
KCs: Kuffer cells.
- •
LPS: lipopolysaccharide.
- •
LXRα: liver X receptor a.
- •
MCP-1: macrophage chemotactic protein 1.
- •
MIP-1: Macrophage Inflammatory Protein 1.
- •
MYD88: myeloid differentiation molecular 88.
- •
NAFL: nonalcoholic fatty liver.
- •
NAFLD: nonalcoholic fatty liver disease.
- •
NASH: nonalcoholic steatohepatitis.
- •
NF-κB: nuclear factor-kappa B.
- •
NKCs: natural killer cells.
- •
SREBP1-c: sterol regulatory element binding protein1-c.
- •
TLRs: Toll-like receptors.
- •
TNF-α: tumor necrosis factor a.
- •
TRAF-6: tumor necrosis factor receptor associated factors 6.
- •
TXB2: thromboxane B2.
- •
XBP-1: X-box binding proteinl.
- •
UCP-2: Uncoupling protein 2.
- •
UPRE: unfolded protein response element.
This review was performed by the Chongqing Key Laboratory of Hepatobiliary Surgery and the Department of Hepatobiliary Surgery of the Second Affiliated Hospital of Chongqing Medical University.
FundingThis review is supported by National Natural Science Foundation of China (NO: 81071339, NO: 31370753).



