




Background: The recurrent microlithiasis represents one of the most frequent clinical forms of lithiasis of the bile ducts. This affection is characterized by the presence of cholesterolic microgallstones on hepatic canaliculars, and belongs to a heterogeneous group of autosomal recessive liver disorders. Radiological diagnosis can be confirmed by analysis of MDR3 gene, coding a protein involved in physiologic translocation of phospholipids in bile. Discovery of MDR3 mutations is of particular interest, since normally associated with good effectiveness of medication by ursodesoxycholic acid. AIM: To review MDR3 mutations in humans associated with recurrent cholesterol microlithiasis and to suggest a practical approach for MDR3 gene analysis. Results: 48 mutations of MDR3 gene have been reported in humans to date, from which 43 (89.5%) in the coding region, and 5 splice site mutations have been associated to cholesterol cholelithiasis. 21 (43.8%) of the 43 precited mutations are located in only 8 exons on 28, near transmembrane or nucleotide binding domains of the protein. From the 22 remaining described mutations, 9 (18.8%) are restricted to exon 14. We suggest therefore to start analysis of MDR3 gene by screening exons 6, 7, 9, 10, 12, 14, 17, 23 and 24 with an appropriate protocol in this diagnosis associated with effective treatment. In conclusion such therapeutic orientation is valuable, since recurrent cholesterolic microlithiasis occurs relatively early in life, and by the fact that recurrence of symptoms may occur despite cholecystectomy, or shock-wave therapy.
The recurrent microlithiasis represents one of the most frequent clinical forms (25% of the cases) of lithiasis of the bile ducts. This affection is characterized by the presence of cholesterolic microgallstones on hepatic canaliculars, and belongs to a heterogeneous group of autosomale recessive liver disorders. It occurs especially in young adults, before the age of forty and appears to be strongly related to metabolic syndrome.1 Prevalence is greater in women and increases considerably with age. In some cases, several genetic abnormalities of phospholipids canalicular gene MDR3 are responsible for occurrence of this pathology.
Diagnosis of recurrent microlithiasis is very often radiological (70 to 80% of cases). Intrahepatic hypere-chogenic foci along the biliary tree may be evidenced by ultrasonography, and hepatic bile composition may be determined by duodenoscopy. In a second time, detection of mutations in MDR3 is of particular interest, since association with good effectiveness of the medical treatment by ursodesoxycholic acid has been demonstrated.2
1. Transport carriers of cholesterol in bileBile is an aqueous solution that contains various lipids: bile salts, phospholipids and cholesterol. At low concentrations, hydrosoluble bile salts are dispersed as monomers. Bile phospholipids are hydrophobic lecithins that organize themselves in vesicles in water. In bile, coexistence of bile salts and phospholipids allowssolubilization of cholesterol as micelles.
If a supersaturation of cholesterol occurs, cholesterol in excess is incorporated in vesicles containing two layers of phospholipids. This condition is necessary but does not lead to lithiasis development per se. Nucleation (or cristallization) occurs when vesicles and micelles concentrate in mucus and agglomerate in liposomes. After solubilisation of phospholipids, cholesterol monohydrate crystals constitute first sediment macroscopically visible (sludge) for calculi formation and growth that is favoured by bile stagnation or impaired motility of gallbladder.
2. Clinical and epidemiological aspectsCholelithiasis is characterized by cholesterol or calcium bilirubinate calculi. Cholesterol cholelithiasis is the most common form of cholelithiasis, associated with calculi of cholesterol in the gallbladder, due to an excess of bile cholesterol compared to solubilizing molecules, namely bile salts and phospholipids.
Taking all ages into account, prevalence of cholesterol cholelithiasis is estimated to be 10 to 20% of the population in industrialized countries. It is more prevalent in women and increases considerably with age. This heterogeneous disease can present as gallstones more than 2 mm in diameter or as microlithiasis, often referred to as biliary sludge. Symptoms are found in only 10% of patients, complications such as acute cholecystitis may be observed in approximately 1-2% of patients, and acute pancreatitis is even rare (less than 2% of patients with gallstones on a period of 20 or 30 years). Other complications are represented by cancer of the gallbladder or migration of calculi in the common bile duct. Treatment for this disease is mainly based on surgery with cholecystectomy or biliary drainage with preoperative or perendoscopic extraction of the common bile duct calculi. Recurrence of symptoms may occur despite cholecystectomy, or shockwave therapy. Low bilirubin contents are observed in about 20% of cases of hepatic lithiasis.3
Various presentations of MDR3 mutations are observed:
- •
Homozygous nonsense mutations in the MDR3 gene are thought to be responsible for progressive familial intrahepatic cholestasis-3 (PFIC type 3), characterized by a chronic cholestasis with high serum GGT levels, a characteristic histologic picture (nonsuppurative inflammatory cholangitis with portal inflammation and ductular proliferation), and liver failure before adulthood.10
- •
Intrahepatic Cholestasis of Pregnancy (ICP) is a liver disorder associated with increased risk of intrauterine fetal death and prematurity. There is an increasing evidence that genetically determined dysfunction in multidrug resistance protein 3 might be a risk factor for ICP development.14
- •
Low phospholipid-associated cholelithiasis is characterized by the association of MDR3 mutations and low biliary phospholipid concentration with symptomatic and recurring cholelithiasis. This syndrome is infrequent and corresponds to a peculiar small subgroup of patients with symptomatic gallstone disease.4
Therefore, at least three human liver diseases are due to a single gene deficiency.
3. Pathogenesis of intrahepatic and gallbladder cholelithiasisSome previous reports suggested that there are hepatic transport and secretion defects of phospholipids (phosphatidylcholine) in the setting of increased cholesterogenesis and decreased bile acid synthesis.2 Phospholipid concentrations are significantly decreased in the hepatic bile issuing from both affected and unaffected bile ducts of hepatic lithiasis patients. The loss of phospholipid secretion appears to be the main cause of the non suppurative destruction cholangitis that is observed in patients with progressive familial intrahepatic cholestasis. This provides an important linkage between a transport defect of phospholipids and the development of cholangiopathies and cholesterol lithiasis. Because phospholipids are main carrier and solvent of cholesterol in hepatic bile, genetic variations in biliary phospholipid metabolism might increase the risk of gallstone formation.3
Many studies have documented ethnic and familial clusters of cholesterol gallstones, indicating that gallstone susceptibility has important genetic components and that cholesterol cholelithiasis might be caused by complex interactions between multiple genes and environment.2 However the pathogenesis of recurrent cholesterolic microlithiasis has not been yet fully elucidated. Reports of familial gallstone disease and the high incidence in the industrialized countries point to genetic factors.5
Rosmorduc et al identified mutations in the ABCB4 (MRD3) gene in 6 symptomatic adult patients with a peculiar form of biliary gallstone disease characterized by intra-hepatic sludge and gallbladder cholesterol gallstones associated with mild chronic cholestasis, recurrence of symptoms after cholecystectomy, and prevention of recurrence by ursodeoxycholate.2 In this study, several lines of evidence suggest that a MDR3 gene defect is involved in this form of cholesterol cholelithiasis. First, the MDR3 protein is the physiologic translocator of phospholipids in bile. Second, phospholipids (mainly phosphatidylcholine) are the main carrier and solvent of biliary cholesterol. Third, all the patients showed evidence of both intrahepatic and gallbladder cholesterol cholelithiasis. Fourth, the bile from 2 patients was supersatured with cholesterol, associated with a low phospholipid concentration. However, bile salt concentrations were not determined in the hepatic biles in this study. Wang and Carey have demonstrated that five crystallization pathways are present in pathophysiologically relevant model biles as functions of increasing lecithin fraction.6 The five crystallization pathways, detection times of liquid and solid cholesterol crystals and the phase boundaries of relevant ternary diagrams are influenced by bile salt hydrophobicity, total lipid concentration and temperature but not by cholesterol saturation index. Low phospholipid concentrations per se do not lead to fast nucleation of cholesterol crystals, and neither does cholesterol supersaturation per se. Rather, the relative phospholipid concentration compared to bile salt concentration (phospholipid/(bile salt + phospholipid) ratio) determines crystallization behavior. It is interesting to note that most cholesterol gallstone patients have a 3-to 4-fold increase in the proportion of deoxycholate conjugates in hepatic and gallbladder biles consistent with more hydrophobic bile salt favoring more rapid cholesterol monohydrate crystallization under pathophysiological conditions.7
Ursodesoxycholic acid was shown to up-regulate the expression of the protein at the canalicular membrane, minimizing the toxicity of the endogenous hydrophobic bile acids and increase the pool of protective hydrophilic bile acids.4
4. Involvement of MDR3 gene in cholelithiasisThe function of the MDR3 gene has been further elucidated through disruption of its murine homologue mdr2. de Vree et al comparing histopathological profile of two patients and that observed in mdr2 (-/-) mice reported a homozygous 7 bp deletion beginning at amino acid 132, and a nonsense mutation in codon 957 (C/T), both introducing a stop codon.10
In addition to an absence of biliary phospholipid, mdr2(-/-) mice had strongly impaired cholesterol secretion.10 Both the decreased expression levels and alteration of subcellular localization of MDR3 in the liver may correlate with impaired biliary phospholipids secretion. These findings taken together with those in mdr2(-/-) mice indicate that the MDR3 gene is a monogenic risk factor for cholesterol microlithiasis.3
Heterozygosity for a mutated MDR3 seems to predispose individuals to cholestasis and/or cholesterol lithiasis when other pathogenetic mechanisms alter transcriptional regulation, intracellular trafficking, or function within the canalicular membrane and other canalicular ABC transporters.3,8
The early occurrence of gallstones in two patients of Rosmorduc’s study might be consistent with a MDR3 dose effect similar to those observed for JAG1 gene expression in some patients with Alagille syndrome.2 There is a nonsense mutation (1327insT) located in exon 12 in the first nucleotide-binding domain (NBD1) resulting in a frame-shift and introducing a stop codon 4 codons downstream, leading to a truncated protein of 446 amino acids. A missense mutation changed an amino acid threonine into a valine in another patient (T175V). This amino acid has recently been shown to be included in a very conserved cluster of 4 amino acids at position 169-172 (TRLT) in the central portion of intracellular loop of the protein required for adenosine triphosphatase activity. Two patients had the S320F mutation, located in exon 9 at the extremity of transmembrane domain (TM5). The substitution changed an amino acid serine, which is very conserved between the human MDR3 gene and its rodent homologues, into an aromatic phenylalanine. Furthermore, this study has shown that the ABC transporters (which include the P-glycopro-tein) are formed by two homologous and symmetric halves, the transmembrane domains 5-6 and 11-12, which participate in drug binding. Indeed most of mutations located within these specific transmembrane domains affect the substrate specificity of P-glycoprotein by modulating the transport, initial binding, or release of some of its substrates. The last mutation was located in exon 26 close to the second adenosine triphosphate-binding domain in the patient. All of these mutations, which differ from those previously published, may alter the biological function of the MDR3 protein and the biliary phospholipid secretion, leading to a high cholesterol-to-phospholipid ratio in bile and thus a high cholesterol saturation index.
The fact that patients of Rosmorduc’s study did not develop severe hepatobiliary disease during childhood may be explained by the residual MDR3 activity that prevents them until biliary phospholipid secretion falls below a critical threshold under the influence of some additional host and/or environmental factors.
5. MDR3 mutationsDiagnosis of cholesterol microlithiasis is done fortuitously during radiological exams for others indications in more than 90% of patients. Ultrasonography is actually the diagnosis exam. More than 40 MDR3 mutations have been identified and described in humans according to literature.
MDR3 mutations in intrahepatic cholestasis of pregnancy
Dixon et al as Jacquemin et al identified heterozygous mutations of the MDR3 gene in patients exhibiting cholestasis only during pregnancy.9,11 Dixon et al described a missense mutation in exon 14 resulting in a disrupted trafficking to the cell surface and subsequent loss of function of the MDR3 protein.9
However, 1712delT mutation identified by Jacquemin was unlikely to play any significant role in obstetric cholestasis in affected Finnish women.12 A third mutation (R144X), again at the heterozygous state, was further identified in this disorder unique to pregnancy.13 Analyzing genetic variability in BSEP (Bile Salt Export Pump) and MDR3 in intrahepatic cholestasis of pregnancy, PauliMagnus et al identified two mutations affecting evolutionarily conserved amino acids (S320F described below and further again identified by Keitel et al and G762E) and four splicing mutations.14,15 An analysis of exon 14 reported three mutations among 80 ICP italian patients.16 Very recently, a new intronic MDR3 mutation c.3486+5G>A resulting in a 54 bp (3465-3518) inframe deletion via cryptic splicing site activation was proved to be an ICP causative locus. Indeed, linkage analysis of the ICP trait versus this intragenic MDR3 variant yielded a LOD score of 2.48, and either stillbirths and symptomatic gallstone disease were more prevalent in heterozygous relatives than in relatives without the mutation (P = 0.00341).17
MDR3 mutations in PFIC3 and low phospholipid associated cholelithiasis
Considering the wide spectrum of MDR3 deficiency, Jacquemin’s study sequence analysis revealed 16 different mutations.18 Three of those 16 mutations have been reported previously.2 In 12 out of 17 patients, the MDR3 defect was characterized on both alleles and on one allele in 5 patients. Six mutations led to a premature truncation of the protein. Five of these 6 mutations were homozygous in 7 patients, and 1 mutation was heterozygous in 1 patient. This last patient was compound heterozygous and had a heterozygous missense mutation on the second allele. The remaining 10 mutations were missense mutations, predicting an amino acid change in presumed important domains of the protein. Out of the 10 missense mutations, 4 were homozygous and 6 heterozygous. In these heterozygous, the mutation on the second allele has not been identified by PCR-SSCP. One mutation was found in transmembrane domain 2, 6, 7, and 12. Two mutations were located in the first Walker A motif and 3 in the first Walker B motif. One mutation was found between the transmembrane domain 6 and the first Walker A motif. In patients who had a homozygous or heterozygous mutations, heterozygosity was confirmed in their parents. These parents were heterozygous for a mutation leading to premature truncation of the protein in 2 cases and for the R652G missense mutation (considered to be a polymorphism) in one case. According to Jacquemin, MDR3 should be associated with FIC1 and BSEP genes in genetic investigation of PFIC.19
In Rosmorduc’s study, sequence analysis of the RT-PCR products from mononuclear cells showed the 4 following mutations described above:2
- •
Three missense mutations:
- -
523 A > G (ACG > GCG; Thr175 > Val) in exon 6, was present as a heterozygous mutation in one patient. This mutation was not detected in 102 chromosomes (corresponding to 51 independent control patients), showing that it did not correspond to a simple polymorphism in the MDR3 gene. However, it was present in the 2 youngest children of this patient.
- -
959C > T (TCC > TTC; Ser320 > Phe) in exon 9, was present at homozygous state in two patients. This mutation was not detected in 126 chromosomes (corresponding to 63 dependent control patients).
- -
3481 C > T (CCC > TCC; Pro1161 > Ser) in exon 26, was present as a homozygous mutation in one patient. This mutation was not detected in 114 chromosomes (corresponding to 57 independent control patients).
- -
- •
A 1-bp insertion at nucleotide 1327 in exon 12 (as described above) was found as heterozygous mutation in two patients. This mutation was not found in another member of the family who was asymptomatic and presented with normal liver enzyme levels. However it was present in the youngest brother of one the two patients. This mutation was not detected in 110 chromosomes (corresponding to 55 independent control patients). In parents of each patient, heterozygous or homozygous mutations were found at least on one of them.
An epidemiological study concerning infantile onset chronic cholestasis in Taiwan showed that only one patient out of 47 had mutation in MDR3 gene, namely a homozygous 719-bp deletion encompassing exon 5 to 9 and leading to protein truncation.20
It is interesting to note that deletions of the MDR3 gene were also found in transmembrane domains of the protein in primary hepatolithiasis, which contrasts with ordinary cholelithiasis, in which stones are usually located in the gallbladder and/or extrahepatic bile ducts:3
- •
A 77-bp deletion at nucleotides 537-613 in exon 7 in TM3, which results in a frameshift at codon 179 and an early stop codon predicting a truncated protein of only 215 amino acids, found as a heterozygous mutation in two of 16 patients.
- •
A 1-bp deletion at nucleotide 1015 in exon 10 in TM 6 was found as a heterozygous mutation in one of those two patients.
- •
A 242-bp deletion at nucleotides 2683-2924 in exons 2223 in TM 11 was found as a heterozygous mutation in the same patient. No other mutations were found in the other 14 patients.
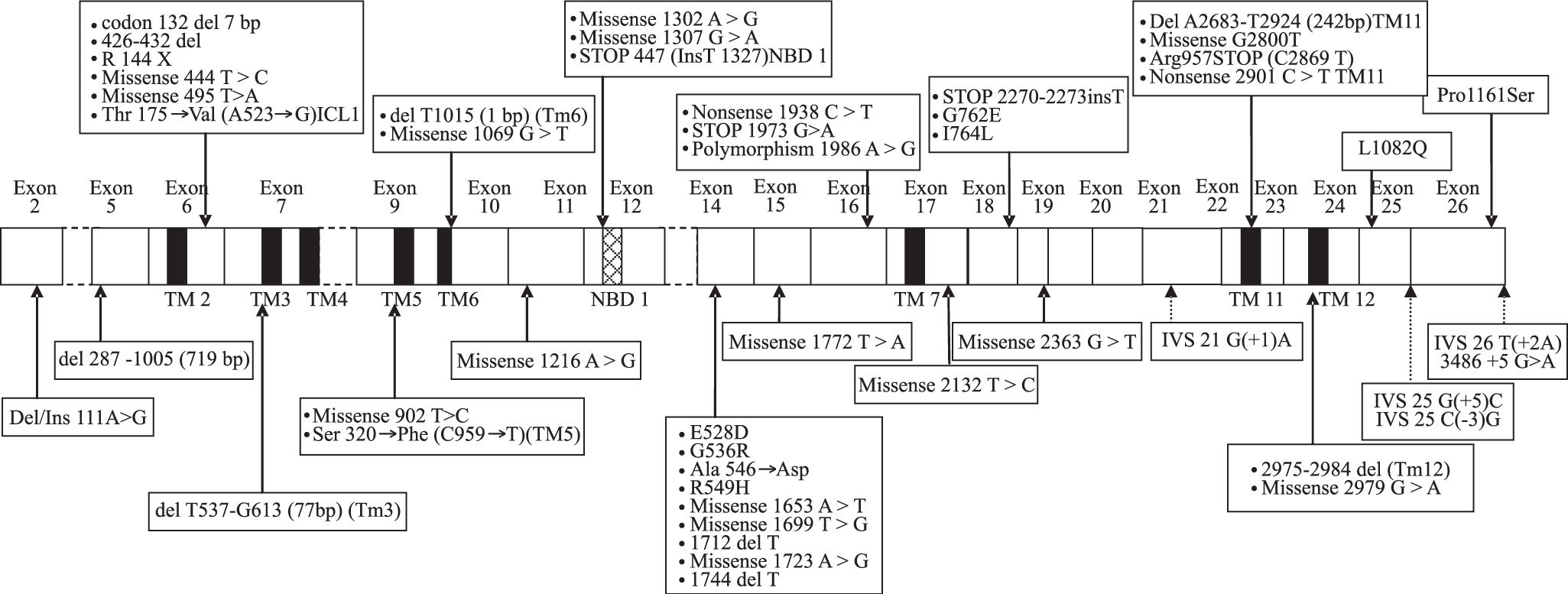
All mutations described above are represented along MDR3 encoding cDNA onFigure 1.
 2 to 6 and the hashed box represents the nucleotide-binding domain (NBD1). The location of the various mutations is represented by arrows (hatched for mutations at splice sites). ICL1 corresponds to the intracellular loop of the MDR3 protein. Mutations located in various domains are in bold in text.")
Various mutations and one polymorphism on the cDNA encoding the MDR3 protein. Exons are represented by white boxes, the black boxes correspond to transmembrane domains (TM) 2 to 6 and the hashed box represents the nucleotide-binding domain (NBD1). The location of the various mutations is represented by arrows (hatched for mutations at splice sites). ICL1 corresponds to the intracellular loop of the MDR3 protein. Mutations located in various domains are in bold in text.
Protocol for MDR3 analysis
To our knowledge, 18 out of 28 exons are affected by mutations associated with recurrent cholesterol microlithiasis. More precisely 48 mutations of MDR3 gene have been reported to date, from which 43 (89.5%) in the coding region. Twenty one (43.8%) are located in only 8 exons near transmembrane or nucleotide binding domains of the protein. From the 22 remaining described mutations, 9 (18.8%) are restricted to exon 14. Lastly, five mutations at splice site have been associated to cholesterol cholelithiasis. In our laboratory we start therefore analysis of MDR3 gene by scanning exons 6, 7, 9, 10, 12, 14, 17, 23 and 24 (corresponding primers are marked with an asterisk inTable I). Focus on this coding region increases chance of mutation discovering, although for any of mutation described above no study was initiated to look for either a founder effect (except for T175V),4 or a hot spot status. Briefly, genomic DNA is isolated from peripheral blood lymphocytes of patient by automated nucleic acids extractor Magnapure™ system (Roche diagnostics, Meylan, France) by standard procedures. PCR amplification of coding exons and flanking intronic regions is carried out using the primer sets as specified inTable I. Amplification of all exons is carried out in 50-pl reaction mixtures containing 50 mM MgCl2, 2 mM DNTP, 25 μM of each primer and 5 Units per microliter of Taq polymerase. All these reagents are from Eurobio (Courtaboeuf, France). After initial denaturation for 7 min at 95°C, amplification is carried out for 36 cycles (30 sec at 95° C, 12 sec at temperature mentioned inTable I and 20 sec at 72°C) and ends by 1 cycle for 7 min at 72° C. PCR products are separated on 1% agarose gel and visualized with ethidium bromide before purification using High Pure PCR Product Purification Kit (Roche Diagnostics, Meylan, France). Sequencing in both sense is carried out in 20-pl mixture containing 1 μL of either 5 μM primer sense or antisens, 8 μl of master Mix (GenomeLab DTCS Quickstart Kit, Beckman Coulter, Villepinte, France) and 1.5 μl of purified DNA. Sequence reaction protocol consists of 30 cycles (20 sec at 96°C, 20 sec at 50°C and 4 min at 60°C), afterwards a precipitation of samples with ethanol is necessary before injection in CEQ 8000TM analyser. Sequences are analyzed by Investigator™ software (Beckman Coulter, Villepinte, France).
Primers used for amplification of MDR3 coding region (exons with an asterisk are those to analyze in priority).
| Exon | Annealing temperature (°C) | Fragment size (bp) | Primer sense; (5' • 3') | Primer antisense: (5' • 3') |
|---|---|---|---|---|
| Exon 2 (88 pb) | 60 | 289 | CAACTTCTTTCCAAGGGTGAAC | CCCCACGTCCCGGGTCTC |
| Exon 3 (55 pb) | 40 | 254 | CTAGACAATCTTAAAGTATTTGCT | GTTTAGTTACAATATTCAACT |
| Exon 4 (151 pb) | 52 | 355 | CCAGATATCCAAATCATTAAATAAG | TCTTTGGATTTTTATTTAAAGTCAG |
| Exon 5 (58 pb) | 52 | 274 | GTGAATCTGGGTAAAGAGTACACGT | AAATG TATGAAAGCTAGAAGATGC |
| *Exon 6 (192 pb) | 50 | 347 | CTTTCCTTGACATATTTTCACAC | TCTGAAACTGAGTCTCACTCTGTC |
| *Exon 7 (172 pb) | 52 | 325 | CAAGTACGAGACTTCTGCATATT | GATGAAGTAGACATCCAACAAACACA |
| Exon 8 (125 pb) | 52 | 303 | AAAAAACACATACCACAAAGAAGAA | CTTTTTCTTCCCAAACCCTCTC |
| *Exon 9 (172 pb) | 58 | 361 | TCATGTTCATCTTTCAAAAAGGAGCG | CGGCTCCTATATAGTTGGAGGTTT |
| *Exon 10 (114 pb) | 52 | 324 | GTAGTTGATTCAAAAATATGCAAAC | AAGTTTAGGTTTATCCTTTCCTTT |
| Exon 11 (111 pb) | 52 | 292 | GAAAAGGCACATAAGTATCAAAA | AGGAATATGATTCAGCAAATTCCA |
| *Exon 12 (126 pb) | 52 | 330 | CAAAACTGGATTCACACGCAA | TGAAAAAAAAGTATCAAATAAGGGA |
| Exon 13 (204 pb) | 52 | 390 | C CTTTGAAGAATAAACTCAGTCC | TTTAACTTGGTTCAGTGTTTAGAAA |
| *Exon 14 (171 pb) | 58 | 352 | TTTCTCAGCCCAGACTCCGGAAG | CATAAAGACAACATGGAGCTTTG |
| Exon 15 (162 pb) | 52 | 337 | TCTTGCTCAGTATAGCATTCACTG | ACATGCACAGTTAAGCACTTGG |
| Exon 16 (171 pb) | 52 | 345 | GCAAGGCTAAGAATTTCAATAATTG | CCTCCAAGTTTAGAATCATAAAGCA |
| *Exon 17 (147 pb) | 58 | 339 | AAGGCTGCTTAATCCCAGAA | GCATGGCCATTGCCCTCAA |
| Exon 18 (105 pb) | 52 | 275 | TTTCACTGTAAGAATTTGGAAGCTC | GAGTGTCACATGTTTAAAACTAGGC |
| Exon 19 (78 pb) | 58 | 313 | ATCCTTGCGTGAATACCCT | GAGTTGTTTTACAGTTGCCACGG |
| Exon 20 (84 pb) | 58 | 249 | CAAGTGTGGGTATGCTACATGCTT | AAAGCTCCAATGCTGAGGAA |
| Exon 21 (204 pb) | 54 | 409 | TTGTAGTGGGCACAAACATTTAA | TTGGTCAGAATGATGAGTGGGT |
| Exon 22 (101 pb) | 54 | 277 | TCTTTTTGGGACAATAATTCAGC | ATCCTGAAGGTTACGTTGATAGGC |
| *Exon 23 (141 pb) | 54 | 332 | CACAGGAGTCATTTTTTTCCTAC | TAAGTCTGGCCGAGTGGGTTTAA |
| *Exon 24 (157 pb) | 52 | 335 | CTGTAGCTATAATCTAGCAGACAGC | CCAGTATTTTTTCCCTTTACTCC |
| Exon 25 (198 pb) | 52 | 388 | GGATTCTAGGTCTACATTTGTCCA | CAAGGAAACTTTACCAAAGACTG |
| Exon 26 (207 pb) | 58 | 350 | CTTTGGTAATTGTTTGGGGG | AAGTTACTAGATTGTCCATTTGGA |
| Exon 27 (147 pb) | 52 | 353 | CCCTGTGCTTGTGTGTGTTTTT | AACAGTTTCTATAACGTTGTTTCAA |
| Exon 28 (258 pb) | 50 | 423 | GGTCTTCTAAATTGATCTAGAATG | TCAAGTAAATTTGAAAATGTTACCT |
Defects of the phospholipid export pump MDR3 result in impaired biliary excretion of phosphatidylcholine and a variety of cholestatic syndromes ranging from progressive familial intrahepatic cholestasis in neonates to biliary cirrhosis in adults.21 Interestingly, great majority of reported mutations affects functional domains of the protein leading to start the analysis near these regions. More specifically, mutations are more frequently localized in symmetrical positions in the protein, in transmembrane domains TM2, TM3, TM4, TM5 and TM6 (around exons 6 to 9) in one side, and TM 11 and TM12 (around exons 23-24) in the other side of the protein. The molecular defects either result in truncation of the open reading frame (frame shift and non-sense) or amino acid substitutions (missense). If frame-shift mutations are associated with severe RNA depletion, most missense mutations are accompanied by faint or absent MDR3 canalicular staining.18 Reduced MDR3 expression can result from messenger RNA destabilization or degradation of a misfolded protein. Functional consequences and resulting phenotype of missense mutations, such as those present in the NBD or Walker motifs, can be predicted with some level of confidence, and their linkage to disease state is uncomplicated. However, mutations in other regions of the coding sequence that do not affect protein expression are more difficult to interpret. In absence of more extensive genetic analysis, it is difficult to conclude that these defects are the sole cause of the disease.
Different mutations in a single gene can generate disparate phenotypes. Loss-of-function mutations that abolish protein expression lead to severe cholestasis in infancy and do not respond to treatment with ursodesoxycholate. Conversely, patients carrying missense mutations, which may permit limited protein expression and function, manifest disease later in life and respond to ursodes-oxycholate.8
Main mutations in the hepatic phospholipid transporter that have been found in ICP patients are missense or splicing mutations at the heterozygous state. Some authors warn against the possibility that other rare underlying hepatic disorders may be unmasked during pregnancy with cholestasis as its first manifestation.22
In a similar way it is noteworthy that I764L and L1082Q mutations that have been mentioned inFigure 1 were involved in specific situations of drug-induced cholestasis and drug-induced hepatocellular injury, respectively. Therefore one should consider these genetically determined functional impairments as predisposition factors rather than causal mutations for intrahepatic cholestasis.23 The mutation screening method used in our laboratory is unable to detect major DNA rearrangements, and does not include the promoter or other potential regulation regions of the gene. Alternatively, defects in other regions of the gene or in other genes may also be involved. If numerous ABC transporters that participate to bile formation and that have been involved in cholesterol cholelithiasis (like Bile Salt Export Pump) have been described, CYP7A1 seems to be a second important actor, since it is the rate-limiting enzyme in the synthesis of bile salts from cholesterol in the liver. Recently, several common DNA polymorphisms in the ABCG8 gene were discovered that are associated with variations in plasma sterols, which could also influence biliary cholesterol secretion, but there is still a paucity of human studies.23
Lastly, it is tempting to speculate that MDR3 defects could also play an important role in cholangiopathies in humans. Indeed, MDR3 variants could play a role as modifier gene in primary biliary cirrhosis and primary sclerosing cholangitis, but their exact role needs further clarification.24
6. ConclusionMultidrug resistance protein 3, as an ABC transporter, participates to hepatic transport and secretion of phospholipid (phosphatidylcholine) and at this account, may be at origin of common biliary cholesterol microlithiasis. Progressive familial intrahepatic cholestasis-3 is a heterogeneous group of autosomal recessive liver disorders characterized by early onset of cholestasis that progresses to hepatic fibrosis, cirrhosis, end-stage liver disease before adulthood. High gamma-glutamyltranspeptidase levels are an additional criterion characteristic to PFIC3 that is necessary to consider analysis of MDR3 gene. In that case, we showed that a preliminary scanning of 8 exons which represents less than the third of the coding region allows detection of main deleterious mutations. Such work is valuable since an effective treatment is now identified. At the same time several drugs for the treatment of cholestatic liver diseases that target MDR3 expression and function are tested, further underscoring the clinical significance of this transport system.
E. Mbongo-Kama and F. Ceppa contributed equally to this work.



