



Background. We have previously reported that Notch signaling pathway protects hepatocytes from ische-mia/reperfusion (I/R) injury by repressing reactive oxygen species (ROS) production. However, apart from hepatocytes, non-parenchymal cells including vascular endothelia cells, Kupffer cells and hepatic stellate cells are also reported to be involved in hepatic I/R injury.
Aim. To clarify the role of Notch signaling in non-parenchymal cells subjected to I/R injury.
Materials and methods. Human Umbilical Vein Endothelial Cells (HUVECs), mouse macrophage line RAW264.7 and rat hepatic stellate cell line HSC-T6 were cultured and subjected to I/R injury, respectively. Activation of Notch signaling was assessed by NICD western blot. Then, pharmacological inhibitor (γ-secretase inhibitor GSI) was used to block Notch signaling of related cell lines in vitro. Intracellular ROS was detected and analyzed by FACS and apoptosis was examined by TUNEL staining and Annexin V staining. Results. Notch signaling responded to I/R injury and I/R injury induced activation of Notch signaling in nonparenchymal cells. Notch signal deficiency led to overproduction of ROS and aggravated cell death of non-parenchymal cells subjected to I/R injury.
Conclusion. Notch signal protectes non-parenchymal cells from I/R injury by repressing ROS.
Organ injury can be incurred by transient ischemia followed by reperfusion, which is a pivotal mechanism of tissue damage in events such as stroke and myocardial infarction and in organ transplantation and vascular surgeries. Hepatic ischemia/reperfusion (I/R) injury can develop during liver transplantation, surgical removal of hepatic tumors, traumas, circulation shock, acute exposure to toxic substances, and other insults.1 Hepatic I/R injury is known to be initiated by the accumulation of reactive oxygen species (ROS).2 ROS impair cells directly through lipid peroxidation, protein oxidation and DNA damage, which together may finally induce cell death. Moreover, ROS and oxidized molecules act as signaling molecules followed by inflammatory responses. + However, molecular mechanisms control ling the ROS accumulation have not been fully elucidated, which hampers effective clinical interference of I/R injury and sometimes lethally exacerbates tissue ischemic damage.
The Notch signaling pathway is highly conserved through evolution and regulates cell proliferation, apoptosis, and cell fate decisions in a broad range of tissues 5. Both Notch receptors and ligands are type I transmembrane proteins mediating direct cell-cell signaling. In mammals, four Notch receptors (Notch1-4) and five ligands (Jagged 1, 2, and Deltalike [Dll] 1, 3, and 4) have been identified. Canonical Notch activation involves receptor cleavage within the transmembrane domain by y-secretase-mediated consecutive enzymatic reactions. This process releases Notch intracellular domain (NICD) that subsequently translocates into the nucleus, where it interacts with the transcription factor C promoter-binding factor 1/recombination signal binding protein Jκ (RBP-J). This protein-protein interaction leads to the dissociation of the RBP-J-centered transcription repression complex and the subsequent formation of a transcription activation complex, including Mastermind-like (MAML) and p300/CBP, which transactivates the transcription of target genes such as the hairy and enhancer of split (Hes) family basic helix-loop-helix (bHLH) factors.6,7
Recent studies have revealed that Notch signaling regulates cell responses to extracellular insults.8–10 Our previous research has suggested that the Notch signaling pathway protects hepatocytes from I/R injury by repressing the production of ROS through JAK2/STAT3 signaling.11 But, how Notch signal works in non-parenchymal cells is still unclear. In the present study, by in vitro experiments we explored preliminarily the role of Notch signaling pathway in endothelia cells, macrophages and hepatic stellate cells in I/R injury.
Materials and MethodsCell cultureThe Human Umbilical Vein Endothelial Cells (HUVECs) (Life technologies, Bei Jing, China) which represented hepatic endothelia cells12 were cultured with RPMI1640 supplemented with 20% fetal bovine serum. Mouse macrophage line RAW264.7 (Zhong Yuan LTD, Bei Jing, China) which represented macrophages localized in liver13 was cultured with RPMI1640 supplemented with 10% fetal bovine serum. And rat hepatic stellate cell line HSC-T6 (Hong Sheng Bio. Shang Hai, China) was cultured with RPMI1640 supplemented with 20% fetal bovine serum. In vitro I/R injury of non-parenchymal cells was performed as described.14 For the cellular hypoxia-reoxygenation (I/R) injury in vitro, HUVECs, RAW264.7 cells or HSC-T6 cells were incubated in Krebs-Henseleit (KH) buffer in a hypoxic chamber (0.5% O2) for 2 h, respectively. Subsequently, hypoxic KH was replaced by normoxic normal medium and were cultured further for 6 h.15 The cells were then collected for further analysis. A γ-secretase inhibitor (GSI IX; Calbiochem, La Jolla, CA) was used at the concentration of 75 juM, with diethyl sulfoxide (DMSO) as a control.8
Flow cytometryFor the measurement of intracellular ROS generation, HUVECs, RAW264.7 cells, or HSC-T6 cells were labeled with 2’,7’-dichlorofluorescein (DCFHDA) (S0033, Beyotime, Haimen, China) following the recommended protocols: intracellular ROS levels were determined by measuring the oxidative conversion of cell permeable DCFH-DA to fluorescent dichlo-rofluorescein (DCF). Cells were incubated with DCFH-DA at 37 oC for 20min. Then DCF fluorescence distribution of 200,000 cells was detected by flow cytometry at an excitation wavelength of 488 nm and at an emission wavelength of 535 nm. The level of intracellular ROS was quantified by using mean fluorescent intensity (MFI), and was statistically compared between groups, as described.16 To detect apoptosis cells which had been subjected to I/R injury in vitro, cells were stained with (APC)-conjugated Annexin V (88-8007, eBioscience, CA) following the recommended protocols,17 and were analyzed by FACS.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-End LabelingThe terminal deoxynucleotidyl transferase-me-diated dUTP nick-end labeling (TUNEL) assay was performed on 5-jum cryostat sections using the TUNEL kit (G3250, Promega, Madison, WI) according to the manufacturer’s protocol. TUNEL-posi tive( + ) cells were detected under light microscopy. Terminal transferase was omitted as a negative control. Positive controls were generated by treatment with DNase 1 (30 U/ml in 40 mmol/L of Tris-Cl, pH 7.6, 6 mmol/L MgCl2, and 2 mmol/L CaCl2 for 30 min).
Western blotCellular protein extracts from cultured cells were prepared with the RIPA lysis buffer (Beyo time, Haimen, China). Protein samples were subjected to SDS-polyacrylamide gel electrophoresis (PAGE), followed by electro-blotting onto PVDF membranes. The membranes were probed with first antibodies including: anti- Notch1 intracellular domain (NICD) (Santa Cruz Biotechnology) or anti-β-actin (Sigma-Aldrich). As secondary antibodies, anti-rabbit-IgG (Boster Bio Tec) or anti-mouse-IgG (Boster Bio Tec) was used. Bands were revealed with enhanced chemiluminescence (ECL; Engreen, Beijing, China).
StatisticsStatistical analysis was performed with the SPSS 12.0 program. Results were expressed as the means ± SD. The comparisons between groups were undertaken using the unpaired Student’s t test. P < 0.05 was considered statistically significant.
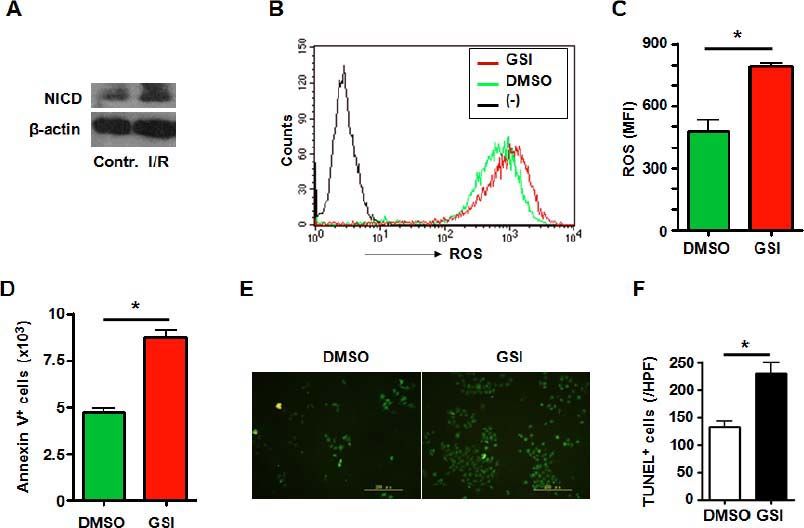
ResultsNotch signal deficiency led to increased ROS accumulation and aggravated cell death in HUVECs subjected to I/R injuryHuman umbilical vein endothelial cells (HUVECs) are cells derived from the endothelium of veins from the umbilical cord. They are used as a laboratory model system for the study of the function and pathology of endothelial cells. We observed role of Notch signaling pathway in vascular endothelia cells subjected to I/R injury. Expression of NICD was increased in the HUVECs suffering I/R injury (Figure 1A), suggesting Notch signal activation during I/R injury. To further clarify the role of Notch signaling pathway in HUVECs subjected to I/R injury, the HUVECs were treated with GSI to block Notch signal (Figure 2A) and were cultured in hypoxic chamber for 2 h followed by normoxic culture for 6 h. We examined ROS level of cultured HUVECs suffering from I/R injury. FACS analysis showed that compared with the control group, blocking Notch signal caused remarkably higher level of ROS accumulation (Figure 1B and C). And Notch signal deficiency led to increased cell death. Annexin V staining showed a significantly increased apoptosis in cells suffering from I/R injury (Figure 1D). TUNEL staining which indicated both apoptosis and necrosis showed increased cell death in GSI treated group (Figures 1E and 1F).
 represents negative control in which cells were not stained with DCFH-DA. C. ROS was quantified by using MFI of each sample. D. HUVECSs cells were subjected to I/R injury in vitro in the presence of DMSO or GSI, and were stained by Annexin V, and 1 x 105 cells of each sample were analyzed by FACS. E. TUNEL. HUVECs were subjected to I/R injury in vitro in the presence of DMSO or GSI, and were stained by TUNEL 6 h after reperfusion. F. Quantification of cell death after TUNEL staining in (E). Five random fields (magnification, x 200) of each sample were counted. Bars = mean ± SD, *P < 0.05.")
A. Notch signal deficiency led to increased ROS accumulation and aggravated cell death in HUVECs subjected to I/R injury. Total cellular proteins were extracted from I/R-injured HUVECs, electrophoresed and blotted, and were detected by using anti-NICD antibody, with β-actin as a reference control. Data represent 3 independent experiments. B. Intracellular ROS level in HUVECs cells analyzed by FACS. HUVECs were treated by I/R injury in vitro in the presence of DMSO or GSI. The production of ROS was examined by FACS. (-) represents negative control in which cells were not stained with DCFH-DA. C. ROS was quantified by using MFI of each sample. D. HUVECSs cells were subjected to I/R injury in vitro in the presence of DMSO or GSI, and were stained by Annexin V, and 1 x 105 cells of each sample were analyzed by FACS. E. TUNEL. HUVECs were subjected to I/R injury in vitro in the presence of DMSO or GSI, and were stained by TUNEL 6 h after reperfusion. F. Quantification of cell death after TUNEL staining in (E). Five random fields (magnification, x 200) of each sample were counted. Bars = mean ± SD, *P < 0.05.
, *P < 0.05, **P < 0.01.")
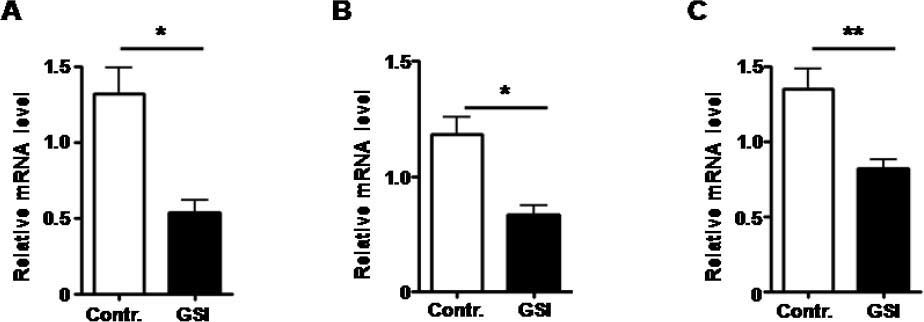
Notch signal was blocked by treated with GSI, as expression of Hesl mRNA was down-regulated. A. The mRNA expression of Hes1 by HUVECs treated by GSI or DMSO. B. The mRNA expression of Hes1 by RAW264.7 cells treated by GSI or DMSO. C. The mRNA expression of Hes1 by HSC-T6 cells treated by GSI or DMSO. Bars = mean ± SD (n = 4), *P < 0.05, **P < 0.01.
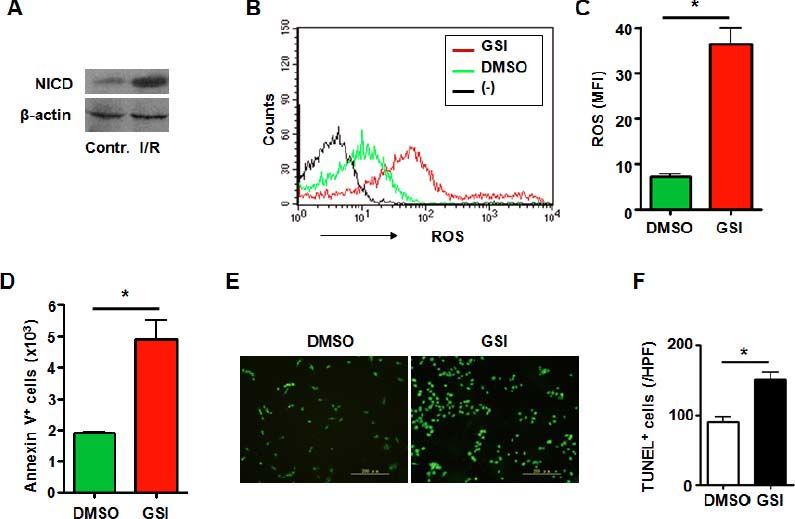
It has been demonstrated that macrophages localizing in liver played an immportant role in process of I/R injury. And Notch signaling has been proved to be a key regulator in macrophages.18 RAW 264.7 cells are a mouse macrophage cell line and is used for research on macrophages. Notch activation was observed in macrophages subjected to I/R injury (Figure 3A). Then we disrupted Notch signaling in mouse macrophage cell line RAW 264.7 (Figure 2B) subjected to I/R injury in vitro. The data showed that increased ROS accumulation was observed in macrophages treated with GSI (Figures 3B and 3C). And like HUVECs, aggravated cell death was detected either by Annexin V staining or TUNEL staining (Figures 3D-3F).
 represents negative control in which cells were not stained with DCFH-DA. C. ROS was quantified by using MFI of each sample. D. RAW264.7 cells were subjected to I/R injury in vitro in the presence of DMSO or GSI, and were stained by Annexin V, and 1 × 105 cells of each sample were analyzed by FACS. E. TUNEL. RAW264.7 cells were subjected to I/R injury in vitro in the presence of DMSO or GSI, and were stained by TUNEL 6 h after reperfusion. F. Quantification of cell death after TUNEL staining in (E). Five random fields (magnification, x 200) of each sample were counted. Bars = mean ± SD, *P < 0.05.")
Notch signal deficiency led to increased ROS accumulation and aggravated cell death in macrophages subjected to I/ R injury. A. Western blot. Total cellular proteins were extracted from I/R-injured RAW264.7 cells, electrophoresed and blotted, and were detected by using anti-NICD antibody, with β-actin as a reference control. Data represent 3 independent experiments. B. Intracellular ROS level in RAW264.7 cells analyzed by FACS. RAW264.7 cells were treated by I/R injury in vitro in the presence of DMSO or GSI. The production of ROS was examined by FACS. (-) represents negative control in which cells were not stained with DCFH-DA. C. ROS was quantified by using MFI of each sample. D. RAW264.7 cells were subjected to I/R injury in vitro in the presence of DMSO or GSI, and were stained by Annexin V, and 1 × 105 cells of each sample were analyzed by FACS. E. TUNEL. RAW264.7 cells were subjected to I/R injury in vitro in the presence of DMSO or GSI, and were stained by TUNEL 6 h after reperfusion. F. Quantification of cell death after TUNEL staining in (E). Five random fields (magnification, x 200) of each sample were counted. Bars = mean ± SD, *P < 0.05.
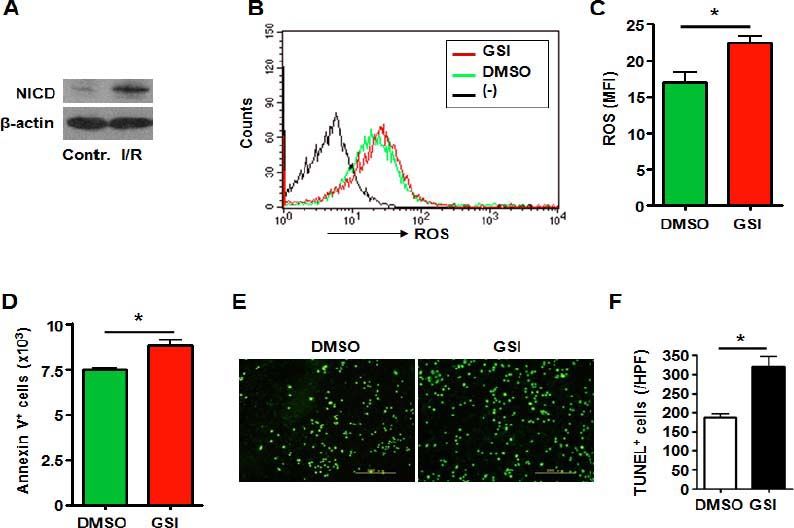
Hepatic stellate cell is another kind of non-parenchymal cell in liver which has been proved to play important role in acute and chronic liver injury.19, 20 Like the HUVECs and RAW264.7 cells, I/R injury caused increased NICD expression in hepatic stel late cell line HSC-T6 suggesting activation of Notch signaling (Figure 4A). And blockade of Notch signaling by treated with GSI (Figure 2C) led to increased ROS accumulation (Figures 4B4C). Increased cell apoptosis was observed in hepatic stellate cells treated with GSI by Annexin V staining (Figure 4D). TUNEL staining showed increased cell death of HSC-T6 caused by Notch deficiency (Figures 4E and 4F).
 represents negative control in which cells were not stained with DCFHDA. C. ROS was quantified by using MFI of each sample. D. HSC-T6 cells were subjected to I/R injury in vitro in the presence of DMSO or GSI, and were stained by Annexin V, and 1 x 105 cells of each sample were analyzed by FACS. E. TUNEL. HSC-T6 cells were subjected to I/R injury in vitro in the presence of DMSO or GSI, and were stained by TUNEL 6 h after reperfusion. F. Quantification of cell death after TUNEL staining in (E). Five random fields (magnification, x 200) of each sample were counted. Bars = mean ± SD, *P < 0.05.")
Notch signal deficiency led to increased ROS accumulation and aggravated cell death in HSC-T6 cells subjected to I/R injury. A. Western blot. Total cellular proteins were extracted from I/R-injured HSC-T6 cells, electrophoresed and blotted, and were detected by using anti-NICD antibody, with β-actin as a reference control. Data represent 3 independent experiments. B. Intracellular ROS level in HSC-T6 cells analyzed by FACS. HSC-T6 cells were treated by I/R injury in vitro in the presence of DMSO or GSI. The production of ROS was examined by FACS. (-) represents negative control in which cells were not stained with DCFHDA. C. ROS was quantified by using MFI of each sample. D. HSC-T6 cells were subjected to I/R injury in vitro in the presence of DMSO or GSI, and were stained by Annexin V, and 1 x 105 cells of each sample were analyzed by FACS. E. TUNEL. HSC-T6 cells were subjected to I/R injury in vitro in the presence of DMSO or GSI, and were stained by TUNEL 6 h after reperfusion. F. Quantification of cell death after TUNEL staining in (E). Five random fields (magnification, x 200) of each sample were counted. Bars = mean ± SD, *P < 0.05.
In previous research we found that Notch signal deficiency caused aggravated hepatic I/R injury. Further we demonstrated that Notch signaling blockade led to increased intracellular ROS accumulation in hepatocytes which caused increased cell death. In addition to hepatocyte liver consists of several types of non-parenchymal cells including vascular endothelia cells, macrophages (Kupffer cells), and hepatic stellate cells, and these non-parenchymal cells also participate in the process of liver injury and reparation.21,22 In the current study, we studied the role of Notch signaling in non-parenchymal cells in the process of I/R injury. Kinds of cell lines of these non-parenchymal cells were treated with GSI or DMSO and were subjected to I/R injury in vitro. The results showed that I/R injury induced activation of Notch signaling and blockade of Notch signaling pathway led to increased ROS accumulation and aggravated cell death in HUVECs, RAW264.7 cells and HSC-T6 cells.
The Notch signaling pathway is highly conserved through evolution and regulates cell proliferation, apoptosis, and cell fate decisions in a broad range of tissues.5 Numerous studies have demonstrated that Notch signaling is critically implicated in the embry onic development and postnatal homeostasis of various tissues.23–25 In recent years, it has also been suggested that Notch signaling participates in cell responses to extra cellular insults. For example, Notch signaling plays a role in the regulatory effects of endothelial cells on hepatocytes during liver regeneration.8 Gude reported that Notch signaling was activated in myocardial I/R and regulated survival of myocardium through the HGF/c-Met and Akt pathway.9 Also it was proved in T-cells that perturbing Notch signaling resulted in accumulation of ROS and reduced expression of anti-apoptotic protein Bcl-xl and finally led to T cell apoptosis.16 And Notch signal regulating intracellular ROS accumulation of hepatocytes in hepatic I/R injury was also demonstrated by us previously.11 In this study we indicated that Notch signaling responded to I/R injury in non-parenchymal cells. As in T cells and hepatocytes, Notch signaling regulated ROS level of vascular endothelia cells, macrophages and hepatic stellate cells. Accaording to our results, I/R injury induced the most obvious ROS increase in vascular endothelia cells among non-parenchymal cells. But GSI led to the most obvious ROS increase in macrophages. In hepatic I/R injury macrophages act as a “murder” which produce ROS to injure the “victims” as hepatocytes and endothelia cells. Notch activation may be a protective mechanism of cell to allevate I/R injury, as regulation of macrphages is more effective. Accumulation of intracellular ROS impairs cells directly through lipid peroxidation, protein oxidation, and DNA damage, which finally induce cell death. Moreover, ROS, as well as the oxidized cellular molecules, act as signaling molecules in various cell types to activate NF-κB and AP-1, which are critical transcription factors governing the following immune responses. In present study we observed increased ROS and increased cell death induced by Notch blockade. Notch signaling regulating intracellular ROS accumulation might be generally applicable. But the molecular mechanisms of Notch signaling regulating ROS in different types of cells are still to be revealed. As ROS plays very important role in kinds of diseases such as ischemia injury, cancer, autoimmune disease and aging, mechanism of Notch regulating ROS may give new clues for research on ROS related diseases.
In hepatic I/R injury in vivo, different types of cells play different role. Vascular endothelia cells and Kupffer cells are activated in the initial phase of I/R injury which induced over production of ROS. And then hepatocytes and hepatic stellate cells are activated and excessive inflammatory response is induced in liver which led to aggravated cell death.2,19,26 And in process of I/R injury in vitro Notch pathway may be a protective signaling which respond to extracellular stimulation. How it works in vivo needs more researches to support.
Abbreviations- •
AP-1: activator protein 1.
- •
bHLH: family basic helix-loop-helix.
- •
DCFH-DA: 2’,7’-dichlorofluorescein.
- •
Dll: Delta-like.
- •
FACS: fluorescence-activated cell sorter.
- •
GSI: y-secretase inhibitor.
- •
Hes: hairy and enhancer of split.
- •
JAK/STAT: Janus kinase/signal transducers and activators of transcription protein.
- •
kI/R: ischemia/reperfusion.
- •
MFI: mean fluorescence intensity.
- •
NF-kB: nuclear factor-κВ.
- •
NICD: Notch intracellular domain.
- •
RBP-J: recombination signal binding protein J.
- •
ROS: reactive oxygen species.
- •
SOCS: suppressor of cytokine signaling.
- •
TUNEL: terminal deoxynucleotidyl transferasemediated dUTP nick-end labeling.
The study was supported by grants from NSFC (81030010), Shan-Xi province fund (2012SF-2-21-2), Xi-Jing fund (XJZT11M01), and CBSKL fund (CBS-KL201201).



