




Primary hepatic neuroendocrine tumors (PHNET) are extremely rare,1,2 which make them challenging both for diagnosis and treatment. Likewise, their classification as primary tumors requires an exhaustive diagnostic study to demonstrate the absence of disease at other levels,3,4 histologic confirmation and prolonged follow-up to rule out the existence of another undiagnosed primary tumor.
We report the case of a patient who was successfully treated with radical surgery that required resection of the hepatic artery (Appleby technique).
The patient is a 71-year-old male who reported epigastric pain radiating toward the right hypochondrium, accompanied by a sensation of postprandial fullness and vomiting in recent hours. Upon examination, he experienced pain during palpation of the epigastrium and right hypochondrium, and a mass was palpated in the epigastrium with hepatomegaly of 2 finger widths.
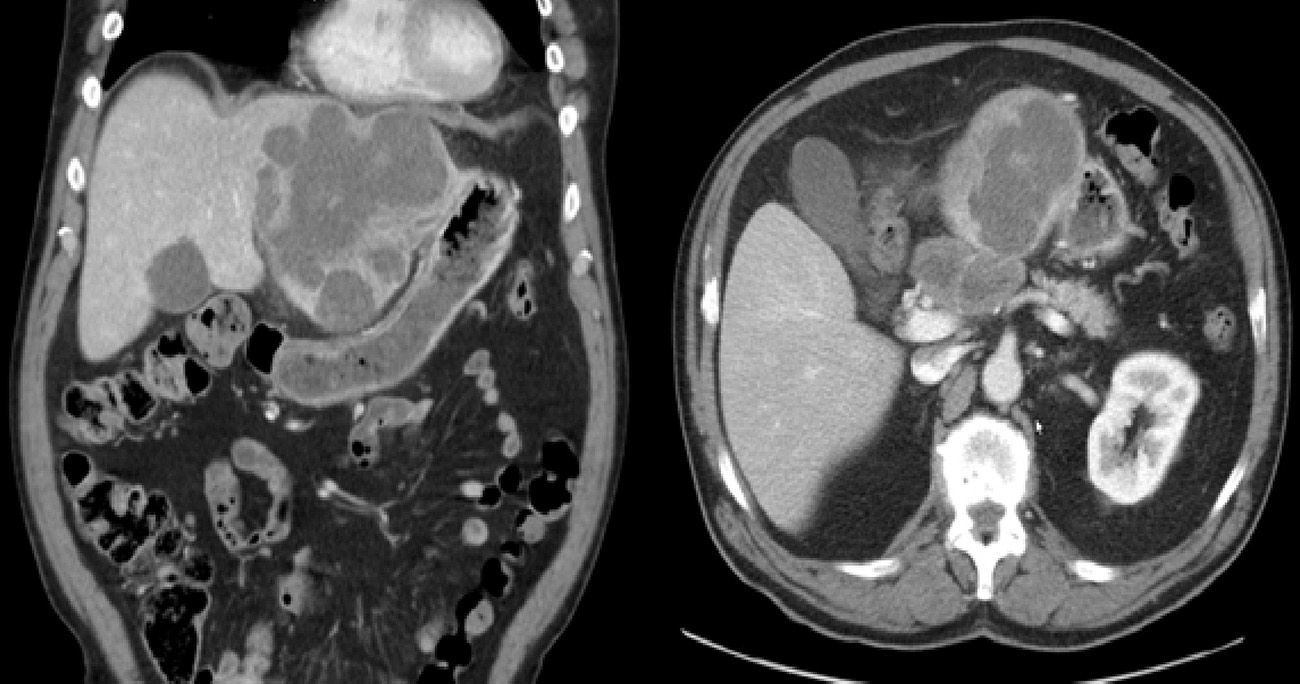
Abdominal ultrasound showed a large solid mass that distended and deformed the left hepatic lobe. CT images were compatible with a polylobed hepatic neoformation, which was probably primary and had uncertain extension to the body of the pancreas and celiac trunk (Fig. 1).

After an extensive study during hospitalization, we ruled out the existence of extrahepatic disease. Tests included upper gastrointestinal endoscopy, colonoscopy, bronchoscopy, MRI, PET-CT and octreoscan with no radiotracer uptake. Lab work-up showed elevated levels of CA 125 62.45U/mL, neurospecific enolase 370ng/mL and chromogranin A 605ng/mL. Gastrin, somatostatin, histamine, glucagon, CEA and AFP levels were normal.
Percutaneous liver biopsy provided histological and immunohistochemical diagnoses that were compatible with neuroendocrine carcinoma.
During surgery, a tumor was observed in segments II–III, and a 6cm lymph-node conglomerate was seen in the celiac trunk with extension to the body of the pancreas. No extrahepatic mass was observed.
We proceeded with segmentectomy II–III and en bloc splenopancreatectomy with the pancreatic body and tail. The lymph-node conglomerate extended toward the celiac trunk and infiltrated the common hepatic artery. After clamping, the proper hepatic artery had adequate flow through the large gastroduodenal artery, and hepatic perfusion was not affected. We therefore decided to perform en bloc resection with the tumor, using no hepatic revascularization (Appleby technique). The patient was discharged on the 9th day post-op, with no further incidents.
The histologic and immunohistochemical studies of the liver and lymph-node conglomerate determined the mass to be a G2 neuroendocrine tumor. The pancreas and spleen were free of relevant histologic alterations.
Eighteen months after the procedure, there was no tumor recurrence.
Neuroendocrine tumors (NET) are comprised of a group of different neoplasms. Their origin is diverse, predominating in the neural crest, thyroid, lungs, endocrine glands, pancreas and gastrointestinal system (54.4%), especially the appendix and terminal ileum. It is therefore important to review previous surgeries, such as the histology of a previous appendectomy, for instance.1,5,6
Growth rates as well as local and general dissemination are relatively low compared to other malignant neoplasms.
The differential diagnosis includes hepatocellular carcinoma, cholangiocarcinoma and metastasis.
There is no evidence of a relationship with cirrhosis or a history of liver disease.7
PHNET can appear at any stage of life, and its most frequent form of appearance is as a single liver mass.1,8 Lymphadenopathies may appear in the celiac trunk in 60% of cases.6
Symptoms are generally nonspecific, and findings are usually incidental. In advanced stages, abdominal pain is related with the mass effect.1
In patients with no chronic liver disease and extrahepatic mass, normal levels of alpha-fetoprotein and cystic changes of the mass, together with diarrhea and abdominal pain (carcinoid syndrome), the diagnosis of functioning PHNET should be considered. In these patients, it is recommended to extend their study with 5-HT, CgA and 5-HIAA tests.1
This syndrome is present in only approximately 5%–10% of cases of PHNET, so they are endocrinologically more silent than extrahepatic carcinoid metastases.6 Gastrin is the most frequently responsible hormone, and chromogranin A is the most useful marker.6,8
Markers AFP, CEA and CA 19.9 lack diagnostic value. Chromogranin A has a sensitivity of 87%–100% and a specificity of 92%, and can also be used as a marker for tumor recurrence.4,5
As for the use of diagnostic biopsies, there is no clear consensus. Postoperative pathology studies are advocated as being the main definitive diagnostic method.4
Octreotide scintigraphy has a specificity close to 83%, thanks to the high affinity of octreotide for somatostatin receptors.1,5 Classic PET-CT with 18-FDG provides little information.
The gold-standard treatment is surgical resection, which provides a 5-year survival that ranges from 74 to 92.5%. Nonetheless, it also has recurrence rates close to 19%, which means that adjuvant therapy should be considered.7,9
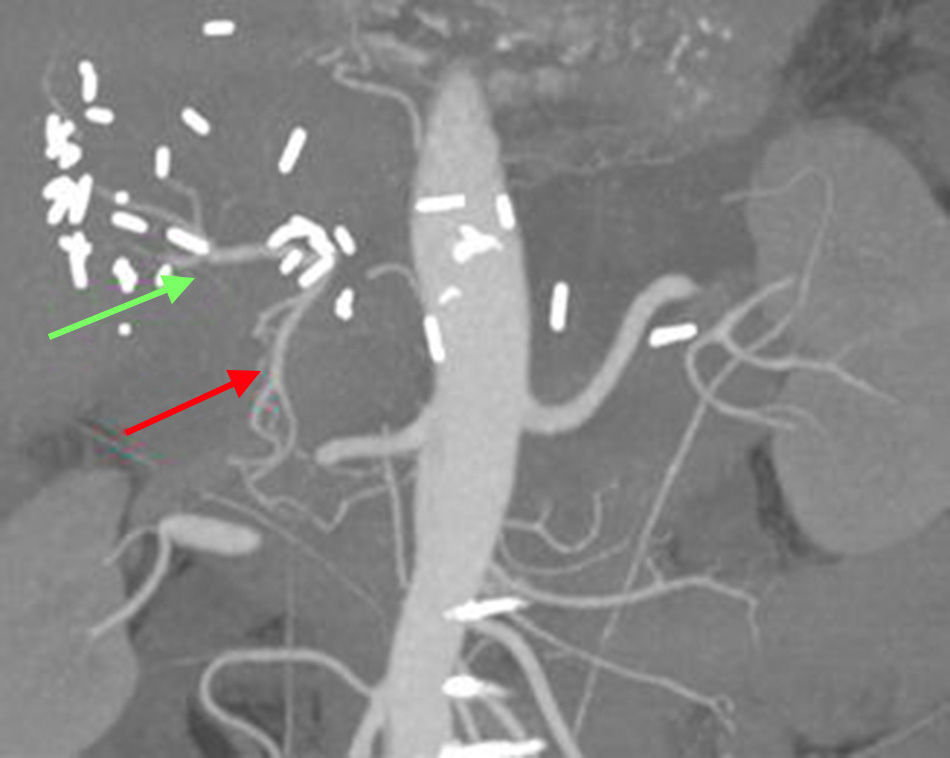
In our patient, the preoperative diagnosis by biopsy allowed us to plan for radical surgery. R0 surgery was performed with resection of the common hepatic artery. This technique, known as the Appleby procedure,10 has been demonstrated to be safe, while increasing survival and ensuring the resectability of locally advanced tumors with vascular invasion. The tumoral obliteration of the common hepatic artery probably favored the vicarious hepatic vascularization through the gastroduodenal artery, as demonstrated by CT angiogram 2 weeks after the procedure (Fig. 2).
 and proper hepatic artery (green).")
In short, although the diagnosis of PHNET is sometimes not early, surgical resection is the best option and provides the most favorable results.
Please cite this article as: Flores García JÁ, Galeano Díaz F, Botello Martínez F, Gallarín Salamanca IM, Blanco Fernández G. Tumor neuroendocrino primario hepático no funcionante con extensión a tronco celiaco. Cir Esp. 2015;93:415–417.
