




El síndrome metabólico (SM) y la hiperlipidemia familiar combinada (HLFC) comparten una parte importante de sus rasgos clínicos, y se ha postulado para ambos una etiología parcialmente coincidente. Se considera que su expresión está afectada por múltiples loci genéticos, existiendo adicionalmente interacción gen-ambiente. En este trabajo se explora la posible implicación en el desarrollo de SM de USF1, un gen previamente asociado con HLFC.
MétodosSe desarrolló un estudio caso-control en el que se analizó la variabilidad genética en USF1 mediante genotipado por pirosecuenciación de los SNP rs2516837, rs2516838, rs6686076, rs2516839, rs2073653, rs2774276, rs2073656, rs2073658 y rs3737787. Se estudiaron 192 sujetos con SM según los criterios ATPIII y 197 sujetos control. Se analizaron las frecuencias alélicas y genotípicas y se estimaron los haplotipos compuestos por los citados polimorfismos. Se analizó estadísticamente la asociación de SNP aislados y haplotipos estimados con el desarrollo de SM.
ResultadosLas frecuencias de los 9 SNP estudiados mostraron que se encontraban en equilibrio de Hardy-Weinberg tanto en casos como en controles. Se apreció un importante grado de desequilibrio de unión entre ellos, pero las diferencias en su distribución entre casos y controles no alcanzaron significación estadística. La estimación de haplotipos de los SNP analizados en los grupos estudiados detectó 6 haplotipos principales, pero ninguno de ellos mostró una asociación estadísticamente significativa con la condición de SM.
ConclusionesEn la muestra de pacientes estudiados no se ha observado una asociación de la variabilidad genética en el locus USF1 con el desarrollo de SM definido según los criterios ATPIII.
Metabolic syndrome (MS) and familial combined hyperlipidemia (FCHL) share many clinical features and a partially overlapping etiology for these two entities has been postulated. The expression of these disorders may be affected by multiple genetic loci in addition to gene-environment interaction. This study explored the possible involvement of the USF1 gene, which has previously been associated with FCHL, in the development of MS.
MethodsA case-control study was performed to analyze genetic variation in USF1 defined by SNPs rs2516837, rs2516838, rs6686076, rs2516839, rs2073653, rs2774276, rs2073656, rs2073658, and rs3737787. SNP genotypes were determined by pyrosequencing in 192 subjects with MS according to the Adult Treatment Panel (ATP)-III criteria and 197 control subjects. Allelic and genotypic frequencies were analyzed for each SNP isolated, and haplotypes were estimated for the combined polymorphisms. A statistical analysis was performed of the association of the SNPs and haplotypes with the development of MS.
ResultsThe frequencies of the nine SNPs studied showed that they were in Hardy-Weinberg equilibrium in both cases and controls. There was a considerable degree of linkage disequilibrium between the SNPs but the differences in their distribution between cases and controls did not reach statistical significance. Estimation of the haplotypes of the SNPs analyzed in the study groups identified six main haplotypes, but none showed a statistically significant association with SM.
ConclusionsIn the sample of patients studied, no association was observed between genetic variation in the USF1 locus and the development of MS defined by ATP-III criteria.
El síndrome metabólico (SM) es un trastorno caracterizado clínicamente por la coexistencia de diferentes factores de riesgo cardiovascular como son la obesidad, la hipertensión, la resistencia a la insulina y la dislipidemia, caracterizada por triglicéridos elevados y colesterol unido a lipoproteínas de alta densidad (c-HDL) disminuido. Estos componentes favorecen el desarrollo de un estado proinflamatorio y protrombótico, y la resultante de todo ello es un riesgo cardiovascular aumentado entre 2 y 3 veces para los sujetos que lo padecen1,2.
El SM es una alteración muy frecuente, con una prevalencia de alrededor del 25% en adultos a partir de 50 años y un incremento asociado a la edad y dependiente del género3–5. El tratamiento de pacientes con SM define un escenario clínico complejo que puede repercutir de forma negativa en su cuidado y hace de su manejo una de las enfermedades más costosas para el sistema sanitario.
El SM comparte ciertas características clínicas con la hiperlipidemia familiar combinada (HLFC), la más común de las dislipidemias hereditarias, con una prevalencia del 1-2% en la población y presente en hasta el 20% de pacientes con enfermedad coronaria prematura6. El rasgo principal de la HLFC es la presencia de un fenotipo lipídico caracterizado por su gran variabilidad entre situaciones de dislipidemia mixta e hipertrigliceridemia o hipercolesterolemia moderadas de manera aislada dentro de una misma familia. Son características fenotípicas de la HLFC una concentración elevada de apoB y un patrón lipoproteico con partículas LDL pequeñas y densas altamente aterogénicas. Además, otros rasgos, como la resistencia a la insulina, la obesidad y la hipertensión, se presentan con gran frecuencia asociados en pacientes que la padecen.
Una posibilidad ampliamente aceptada para la explicación fisiopatológica de la HLFC es que se deriva de una resistencia periférica a la insulina, que puede resultar exacerbada por una situación de obesidad y que podría justificar el fenotipo lipídico mediado por un metabolismo anormal de ácidos grasos, cuya concentración en plasma está elevada, y que sería causante de una hiperproducción de VLDL, resolviéndose en distintos fenotipos lipídicos según la eficiencia lipolítica del individuo7.
Por tanto, el SM y la HLFC son dos entidades clínicas cuyas definiciones se solapan en ciertos aspectos y constituyen fenotipos complejos a los que contribuyen factores genéticos y ambientales. Además, la coincidencia parcial de rasgos entre uno y otro sugiere una etiología que puede compartir factores genéticos básicos, cuyo desarrollo hacia un fenotipo u otro puede estar determinado por factores ambientales u otros factores genéticos modificadores.
En 2004, Pajukanta et al8 identificaron el gen USF1 (upstream stimulatory factor 1) como un gen asociado a la HLFC en una población finlandesa relativamente aislada. Este gen está situado en la región q21 del cromosoma 1, que había sido previamente señalada en distintos análisis genómicos independientes como portadora de un determinante genético de HLFC9,10. Asimismo, un estudio realizado sobre sujetos sanos del estudio EARSII mostró la influencia de este gen en parámetros relacionados con el metabolismo de la glucosa y los lípidos11 y, más recientemente, se ha mostrado su influencia directa en los lípidos plasmáticos y en rasgos metabólicos en un modelo murino12.
Pajukanta et al8 estudiaron la asociación de 9 posiciones nucleotídicas simples polimórficas (SNP: single nucleotide polymorphisms) situadas en posiciones intrónicas y en regiones no codificantes del gen USF1 y pusieron de manifiesto la existencia de un haplotipo protector y otro de predisposición formados por sólo 2 SNP (rs3737787, rs2073658) para el desarrollo de rasgos de la dislipidemia. El perfil de expresión génica en tejido adiposo es diferente para portadores de cada haplotipo, lo cual sugeriría un defecto a nivel básico que podría ser subyacente a las manifestaciones observadas en la HLFC.
El gen USF1 codifica para un factor de transcripción expresado ubicuamente, y que se encuentra implicado en la respuesta del organismo a la glucosa y a la ingesta de hidratos de carbono, y entre los genes que es capaz de activar se incluyen los de varias apolipoproteínas (apoCIII, apoAII, apoE), lipasa hormona-sensible, ácido graso sintasa, glucocinasa, receptor del glucagón, ABCA1 y renina8,13–18. Esto lo sitúa en una posición clave dentro de la red de control transcripcional que regula los procesos que se encuentran perturbados en situaciones de resistencia a la insulina, dislipidemia o hipertensión.
En el presente trabajo se plantea la hipótesis de que la variabilidad genética de USF1 interviene en el desarrollo del SM. Dada la función reconocida de USF1 en el control del metabolismo de los hidratos de carbono y la síntesis de ácidos grasos, así como la relación existente a nivel fenotípico entre la HLFC y el SM, resulta plausible la implicación de variantes alélicas comunes en el locus USF1 en la etiología del SM. Para verificar esta hipótesis hemos realizado un estudio caso-control en el que se comparan polimorfismos tipo SNP del gen USF1 en un grupo de pacientes con SM y otro de sujetos sanos.
Materiales y métodosSujetos de estudioEl presente estudio fue aprobado por el Comité de Ética en Investigación Clínica de Aragón (CEICA) y todos los sujetos estudiados dieron su consentimiento informado previamente a su participación. Se obtuvo la colaboración de 192 casos con edades entre 20 y 80 años utilizando los criterios de definición de SM propuestos por el NCEP ATPIII2, siendo criterios de exclusión el tratamiento con fármacos hipolipemiantes, el consumo de alcohol superior a 30g diarios y la coexistencia de enfermedades crónicas de tipo renal, hepático o cáncer. Igualmente se obtuvo la participación de 197 controles con edades entre 20 y 80 años, no sometidos a ningún tratamiento farmacológico y que no presentaran más de un criterio de la definición de SM previamente citada. Para todos los participantes se realizaron determinaciones antropométricas y físicas en el momento de la exploración médica y se obtuvieron muestras de sangre en ayunas que permitieron la realización de medidas bioquímicas por métodos estandarizados, así como la obtención de ADN en un sistema de extracción automática de ADN genómico AutogenFlex 3000 (Autogen) y reactivos FlexiGene DNA AGF3000 (QIAGEN).
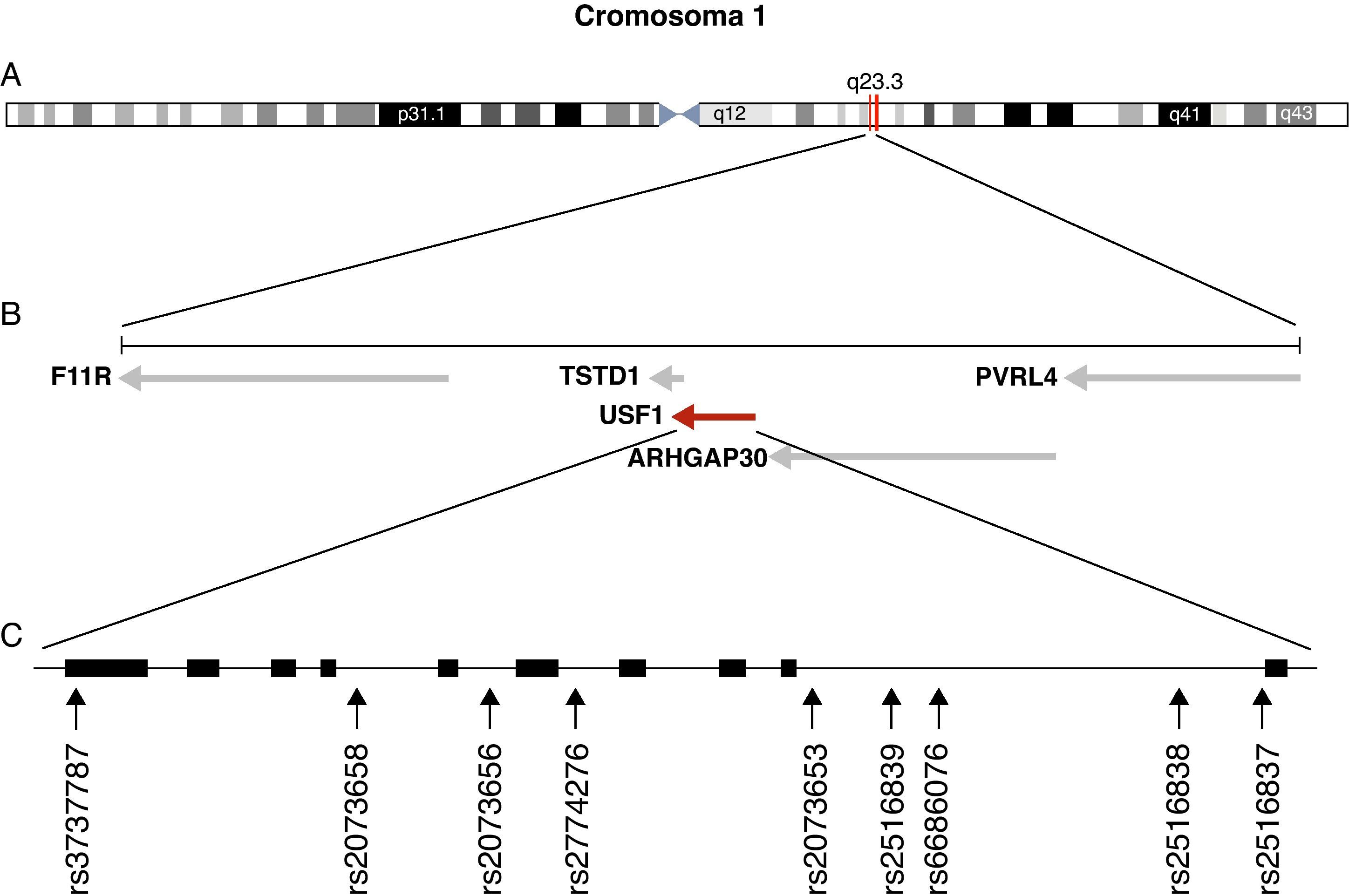
GenotipadoSe seleccionaron 9 SNP distribuidos a lo largo de la extensión del gen USF1 para determinar su variabilidad genética en los grupos de estudio. Los criterios utilizados para su inclusión fueron su existencia en la base de datos dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/) con frecuencias alélicas descritas en poblaciones europeas superiores a 0,1 y que no fueran iguales entre sí para incrementar la probabilidad de que se comportasen como tag SNP. Entre los 9 SNP elegidos para su genotipado en la muestra estudiada se forzó la inclusión de los SNP rs3737787 y rs2073658, para los que Pajukanta et al8 describieron su asociación con HLFC e hipertrigliceridemia. Además de estos dos SNP se analizaron los siguientes: rs2073656, rs2774276, rs2073653, rs2516839, rs6686076, rs2516838 y 2516837. La figura 1 muestra la distribución de los SNP analizados a lo largo del gen USF1.
 Situación del locus USF1 en el cromosoma 1. B) Entorno genómico del gen USF1 en la región 1q23.1. C) Posición relativa de los SNP en el gen USF1.")
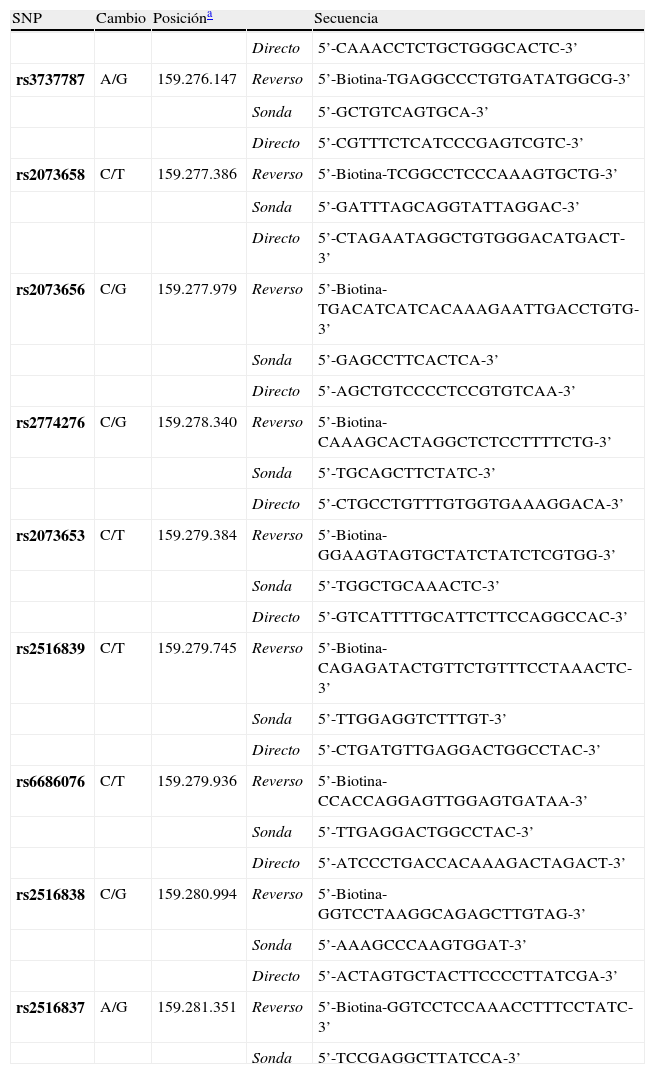
El genotipado para cada SNP en las muestras de ADN genómico de cada participante en el estudio se realizó mediante la técnica de pirosecuenciación19 utilizando un equipo PSQ 96MA (Biotage). Para ello se amplificaron mediante PCR pequeñas secuencias en torno a las posiciones polimórficas utilizando oligonucleótidos específicos, uno de ellos biotinilado en posición 5’ para lograr la inmovilización de la hebra simple de ADN que fue analizada subsiguientemente mediante una sonda adyacente a la posición polimórfica que actúa como cebador en la reacción de pirosecuenciación. Las secuencias de oligonucleótidos utilizadas para el análisis de cada SNP se muestran en la tabla 1. Todas ellas fueron obtenidas del servidor SOP320, excepto las correspondientes a los polimorfismos rs2073658, rs6686076, rs2516838 y rs2516837, que no dieron resultados satisfactorios en el proceso de optimización y fueron sustituidas por nuevos oligonucleótidos diseñados con la ayuda del programa PyroMark Assay Design Software 2.0 (Biotage).
Secuencias de los oligonucleótidos utilizados en el genotipado
| SNP | Cambio | Posicióna | Secuencia | |
| Directo | 5’-CAAACCTCTGCTGGGCACTC-3’ | |||
| rs3737787 | A/G | 159.276.147 | Reverso | 5’-Biotina-TGAGGCCCTGTGATATGGCG-3’ |
| Sonda | 5’-GCTGTCAGTGCA-3’ | |||
| Directo | 5’-CGTTTCTCATCCCGAGTCGTC-3’ | |||
| rs2073658 | C/T | 159.277.386 | Reverso | 5’-Biotina-TCGGCCTCCCAAAGTGCTG-3’ |
| Sonda | 5’-GATTTAGCAGGTATTAGGAC-3’ | |||
| Directo | 5’-CTAGAATAGGCTGTGGGACATGACT-3’ | |||
| rs2073656 | C/G | 159.277.979 | Reverso | 5’-Biotina-TGACATCATCACAAAGAATTGACCTGTG-3’ |
| Sonda | 5’-GAGCCTTCACTCA-3’ | |||
| Directo | 5’-AGCTGTCCCCTCCGTGTCAA-3’ | |||
| rs2774276 | C/G | 159.278.340 | Reverso | 5’-Biotina-CAAAGCACTAGGCTCTCCTTTTCTG-3’ |
| Sonda | 5’-TGCAGCTTCTATC-3’ | |||
| Directo | 5’-CTGCCTGTTTGTGGTGAAAGGACA-3’ | |||
| rs2073653 | C/T | 159.279.384 | Reverso | 5’-Biotina-GGAAGTAGTGCTATCTATCTCGTGG-3’ |
| Sonda | 5’-TGGCTGCAAACTC-3’ | |||
| Directo | 5’-GTCATTTTGCATTCTTCCAGGCCAC-3’ | |||
| rs2516839 | C/T | 159.279.745 | Reverso | 5’-Biotina-CAGAGATACTGTTCTGTTTCCTAAACTC-3’ |
| Sonda | 5’-TTGGAGGTCTTTGT-3’ | |||
| Directo | 5’-CTGATGTTGAGGACTGGCCTAC-3’ | |||
| rs6686076 | C/T | 159.279.936 | Reverso | 5’-Biotina-CCACCAGGAGTTGGAGTGATAA-3’ |
| Sonda | 5’-TTGAGGACTGGCCTAC-3’ | |||
| Directo | 5’-ATCCCTGACCACAAAGACTAGACT-3’ | |||
| rs2516838 | C/G | 159.280.994 | Reverso | 5’-Biotina-GGTCCTAAGGCAGAGCTTGTAG-3’ |
| Sonda | 5’-AAAGCCCAAGTGGAT-3’ | |||
| Directo | 5’-ACTAGTGCTACTTCCCCTTATCGA-3’ | |||
| rs2516837 | A/G | 159.281.351 | Reverso | 5’-Biotina-GGTCCTCCAAACCTTTCCTATC-3’ |
| Sonda | 5’-TCCGAGGCTTATCCA-3’ |
El volumen muestral diseñado para el presente estudio caso-control permitiría una potencia estadística mayor del 70% para un nivel de significación estadística fijado en el 95% en el caso de que las diferencias alélicas estuvieran en un rango similar al descrito por Pajukanta et al8 según estimación realizada utilizando el programa PAWE21.
Análisis estadísticosDatos de variables continuas son presentados como media±desviación estándar o medianas (rango intercuartiles) para aquellos casos en los que no se ajustaban a una distribución normal según el test de Kolmogorov-Smirnoff (p<0,05). Datos de variables categóricas se muestran como número (porcentaje). Diferencias entre variables continuas fueron evaluadas utilizando las pruebas t-test desapareado o de Mann-Whitney, según fuera apropiado, y diferencias en variables categóricas se analizaron mediante la prueba de χ2. Se utilizó el programa SPSS 15.0 (SPSS).
Se analizó si los genotipos aislados para cada SNP estudiado estaban en equilibrio de Hardy-Weinberg mediante una prueba χ2 con un grado de libertad y nivel de significación p<0,05, y se utilizó el programa Haploview 4.122 para estudiar el desequilibrio de unión existente entre los SNP estudiados, tanto utilizando el parámetro D’ (desequilibrio de unión normalizado) como el coeficiente de correlación entre SNP (R2) y para determinar si los SNP estudiados se comportan como tag SNP y determinar posibles bloques haplotípicos.
Se analizó la posible asociación de cada SNP con el desarrollo de SM a nivel de frecuencias alélicas y genotípicas mediante pruebas de χ2 con grados de libertad 1 y 2 respectivamente y un nivel de significación p<0,0056 considerando la corrección de Bonferroni para comparaciones múltiples.
Se utilizó el programa PHASE 2.123,24 para estimar los haplotipos compuestos por los SNP estudiados y realizar un análisis de asociación caso-control para los haplotipos inferidos.
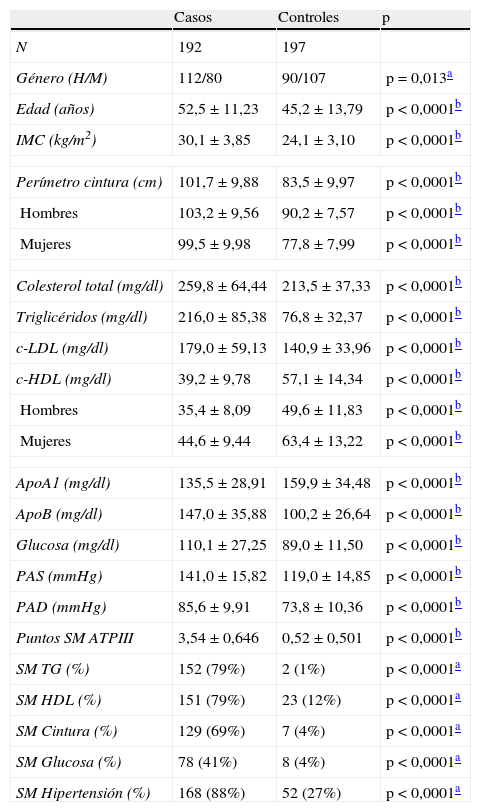
ResultadosPara el desarrollo del estudio caso-control se consiguió la participación de 192 casos y 197 controles. En la tabla 2 se muestran las características antropométricas y clínicas de los sujetos de estudio. Existe un desequilibrio estadísticamente significativo en la distribución por género dentro de cada grupo, habiendo una mayor proporción de hombres respecto a mujeres en el grupo de casos, y viceversa en el grupo de controles. Existen diferencias significativas en el promedio de edad para cada grupo, siendo mayor para el grupo de casos. En el resto de variables analizadas se observan diferencias estadísticamente significativas derivadas de los criterios de definición utilizados para incluir a los participantes en cada grupo.
Características antropométricas y clínicas de casos y controles
| Casos | Controles | p | |
| N | 192 | 197 | |
| Género (H/M) | 112/80 | 90/107 | p=0,013a |
| Edad (años) | 52,5±11,23 | 45,2±13,79 | p<0,0001b |
| IMC (kg/m2) | 30,1±3,85 | 24,1±3,10 | p<0,0001b |
| Perímetro cintura (cm) | 101,7±9,88 | 83,5±9,97 | p<0,0001b |
| Hombres | 103,2±9,56 | 90,2±7,57 | p<0,0001b |
| Mujeres | 99,5±9,98 | 77,8±7,99 | p<0,0001b |
| Colesterol total (mg/dl) | 259,8±64,44 | 213,5±37,33 | p<0,0001b |
| Triglicéridos (mg/dl) | 216,0±85,38 | 76,8±32,37 | p<0,0001b |
| c-LDL (mg/dl) | 179,0±59,13 | 140,9±33,96 | p<0,0001b |
| c-HDL (mg/dl) | 39,2±9,78 | 57,1±14,34 | p<0,0001b |
| Hombres | 35,4±8,09 | 49,6±11,83 | p<0,0001b |
| Mujeres | 44,6±9,44 | 63,4±13,22 | p<0,0001b |
| ApoA1 (mg/dl) | 135,5±28,91 | 159,9±34,48 | p<0,0001b |
| ApoB (mg/dl) | 147,0±35,88 | 100,2±26,64 | p<0,0001b |
| Glucosa (mg/dl) | 110,1±27,25 | 89,0±11,50 | p<0,0001b |
| PAS (mmHg) | 141,0±15,82 | 119,0±14,85 | p<0,0001b |
| PAD (mmHg) | 85,6±9,91 | 73,8±10,36 | p<0,0001b |
| Puntos SM ATPIII | 3,54±0,646 | 0,52±0,501 | p<0,0001b |
| SM TG (%) | 152 (79%) | 2 (1%) | p<0,0001a |
| SM HDL (%) | 151 (79%) | 23 (12%) | p<0,0001a |
| SM Cintura (%) | 129 (69%) | 7 (4%) | p<0,0001a |
| SM Glucosa (%) | 78 (41%) | 8 (4%) | p<0,0001a |
| SM Hipertensión (%) | 168 (88%) | 52 (27%) | p<0,0001a |
En el grupo de casos el criterio de definición de SM más prevalente fue la hipertensión arterial (88%), seguido de la condición dislipidémica (79% igual para hipertrigliceridemia y bajo c-HDL) y perímetro de cintura (69%), siendo la hiperglucemia en ayunas el rasgo menos prevalente (41%). En el grupo de controles sólo dos rasgos asociados al SM alcanzaron una prevalencia superior al 10%: la hipertensión arterial (27%) y la baja concentración de c-HDL (12%).
Se diseñaron ensayos de genotipado basados en la técnica de pirosecuenciación para cada uno de los SNP seleccionados. En la tabla 1 se muestran los oligonucleótidos cebadores que permiten la amplificación por PCR de fragmentos de ADN que contienen cada posición polimórfica y las secuencias de los oligonucleótidos sonda que permiten la determinación de cada polimorfismo mediante la obtención de patrones inequívocos de pirosecuenciación. Esta técnica permitió analizar un polimorfismo en 96 muestras al día una vez optimizadas las condiciones del ensayo.
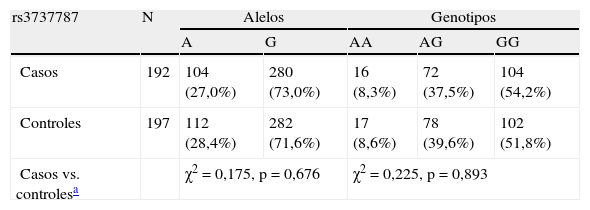
Se calcularon las frecuencias genotípicas y, a partir de ellas, las frecuencias alélicas. Ambas se describen en la tabla 3 para cada polimorfismo considerado aisladamente. Las frecuencias genotípicas de todos los polimorfismos se encontraron en equilibrio de Hardy-Weinberg. Las frecuencias obtenidas para la muestra de población estudiada muestran rangos similares a las descritas en bases de datos públicas (dbSNP en NCBI, http://www.ncbi.nlm.nih.gov/projects/SNP/index.html) para poblaciones de origen europeo, aunque no se corresponden exactamente con ellas tal como cabe esperar de las diferencias existentes entre la población española y las muestras de referencia europeas extraídas de población centroeuropea.
Frecuencias de los SNP estudiados en casos y controles
| rs3737787 | N | Alelos | Genotipos | |||
| A | G | AA | AG | GG | ||
| Casos | 192 | 104 (27,0%) | 280 (73,0%) | 16 (8,3%) | 72 (37,5%) | 104 (54,2%) |
| Controles | 197 | 112 (28,4%) | 282 (71,6%) | 17 (8,6%) | 78 (39,6%) | 102 (51,8%) |
| Casos vs. controlesa | χ2=0,175, p=0,676 | χ2=0,225, p=0,893 | ||||
| rs2073658 | N | Alelos | Genotipos | |||
| C | T | CC | CT | TT | ||
| Casos | 192 | 278 (72,4%) | 106 (27,6%) | 104 (54,2%) | 70 (36,5%) | 18 (9,4%) |
| Controles | 197 | 282 (71,6%) | 112 (28,4%) | 103 (52,3%) | 76 (38,6%) | 18 (9,1%) |
| Casos vs. controlesa | χ2=0,065, p=0,798 | χ2=0,187, p=0,911 | ||||
| rs2073656 | N | Alelos | Genotipos | |||
| C | G | CC | CG | GG | ||
| Casos | 192 | 281 (73,20%) | 103 (26,8%) | 105 (54,7%) | 71 (37,0%) | 16 (8,3%) |
| Controles | 197 | 282 (71,6%) | 112 (28,4%) | 102 (51,8%) | 78 (39,6%) | 17 (8,6%) |
| Casos vs. controlesa | χ2=0,250, p=0,617 | χ2=338, p=0,844 | ||||
| rs2774276 | N | Alelos | Genotipos | |||
| C | G | CC | CG | GG | ||
| Casos | 192 | 284 (74,0%) | 100 (26,0%) | 105 (54,7%) | 74 (38,5%) | 13 (6,8%) |
| Controles | 197 | 293 (74,4%) | 101 (25,6%) | 112 (56,9%) | 69 (35,0%) | 16 (8,1%) |
| Casos vs. controlesa | χ2=0,017, p=0,897 | χ2=647, p=0,724 | ||||
| rs2073653 | N | Alelos | Genotipos | |||
| C | T | CC | CT | TT | ||
| Casos | 192 | 38 (9,9%) | 346 (90,1%) | 3 (1,6%) | 32 (16,7%) | 157 (81,8%) |
| Controles | 197 | 35 (8,8%) | 359 (91,2%) | 3 (1,5%) | 29 (14,7%) | 165 (83,8%) |
| Casos vs. controlesa | χ2=0,235, p=0,628 | χ2=282, p=868 | ||||
| rs2516839 | N | Alelos | Genotipos | |||
| C | T | CC | CT | TT | ||
| Casos | 192 | 139 (36,2%) | 245 (63,8%) | 27 (14,1%) | 85 (44,3%) | 80 (41,7%) |
| Controles | 197 | 135 (34,2%) | 259 (65,78%) | 27 (13,7%) | 81 (41,1%) | 89 (45,2%) |
| Casos vs. controlesa | χ2=0,319, p=0,572 | χ2=511, p=0,774 | ||||
| rs6686076 | N | Alelos | Genotipos | |||
| C | T | CC | CT | TT | ||
| Casos | 192 | 38 (9,9%) | 346 (90,1%) | 3 (1,6%) | 32 (16,7%) | 157 (81,8%) |
| Controles | 197 | 35 (8,8%) | 359 (91,2%) | 3 (1,5%) | 29 (14,7%) | 165 (83,8%) |
| Casos vs. controlesa | χ2=0,235, p=0,628 | χ2=282, p=0,868 | ||||
| rs2516838 | N | Alelos | Genotipos | |||
| C | G | CC | CG | GG | ||
| Casos | 192 | 244 (63,5%) | 140 (36,5%) | 75 (39,1%) | 94 (49,0%) | 23 (12,0%) |
| Controles | 197 | 246 (62,5%) | 148 (37,5%) | 79 (40,1%) | 88 (44,7%) | 30 (15,2%) |
| Casos vs. controlesa | χ2=0,102, p=0,750 | χ2=1,162, p=0,559 | ||||
| rs2516837 | N | Alelos | Genotipos | |||
| A | G | AA | AG | GG | ||
| Casos | 192 | 110 (28,7%) | 274 (71,3%) | 12 (6,3%) | 86 (44,8%) | 94 (49,0%) |
| Controles | 197 | 100 (25,4%) | 294 (74,6%) | 12 (6,1%) | 76 (38,6%) | 109 (55,3%) |
| Casos vs. controlesa | χ2=1,052, p=0,3050 | χ2=1,662, p=0,436 | ||||
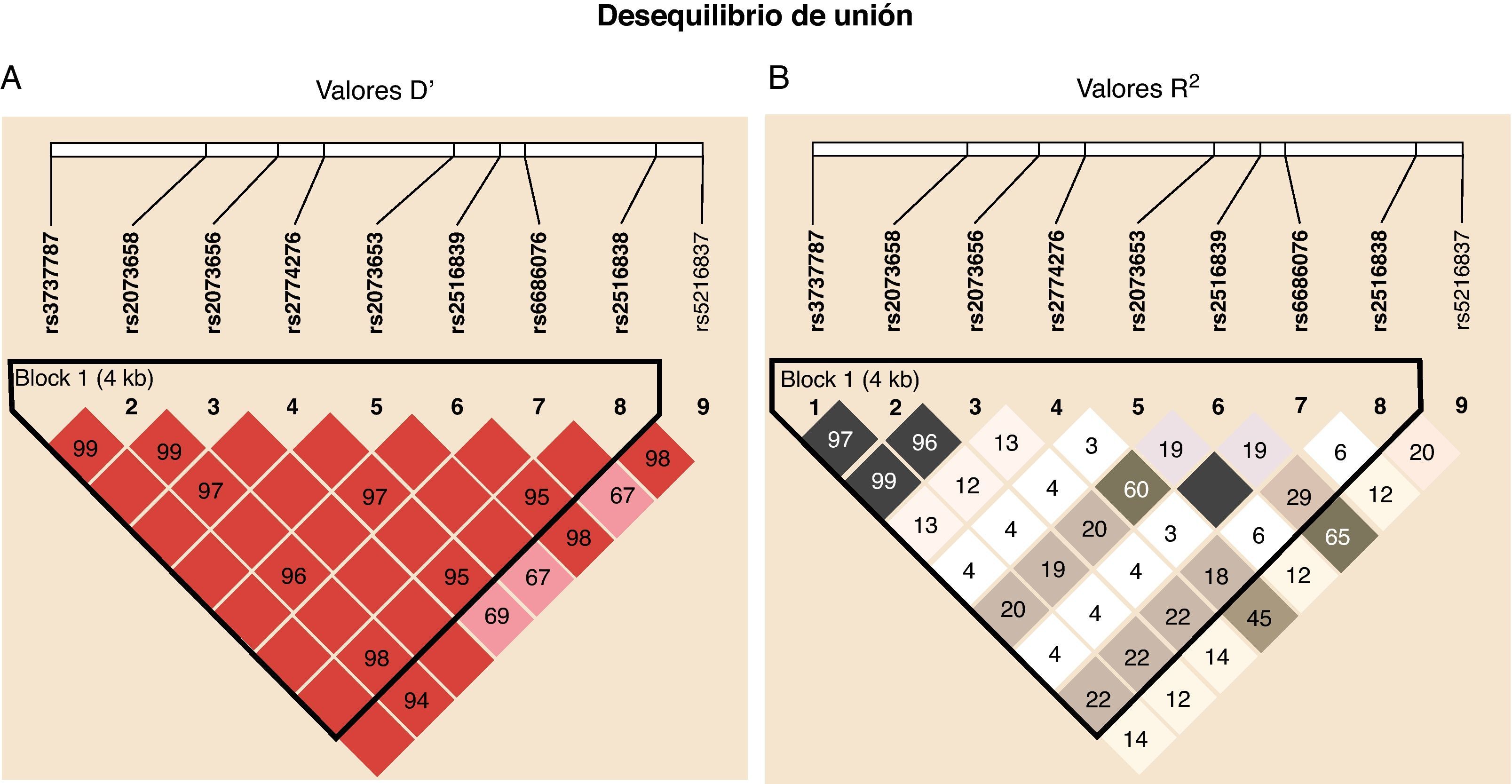
El análisis de los datos de genotipado con el programa Haploview proporcionó información en dos aspectos diferentes: por una parte mostró que entre los SNP estudiados en USF1 existe un fuerte desequilibrio de unión, según los resultados del parámetro D’ (desequilibrio de unión normalizado), que fue 1 en la mayor parte de los emparejamientos de SNP y muy alto en los demás (fig. 2A), aunque el parámetro R2 (correlación entre alelos) mostrado en la figura 2B resultó parcialmente contradictorio al no alcanzar valores tan altos, aunque claramente sugerentes de la existencia de un ligamiento entre polimorfismos. Este aparente desacuerdo entre medidas del desequilibrio de unión puede deberse a diferentes factores que afectan al desequilibrio de unión, como es el caso del ratio de mutación en el locus que puede dar lugar a diversidad preferentemente respecto a fenómenos de recombinación25. En base a estos resultados, y utilizando el algoritmo de Gabriel26 con los valores instaurados por defecto que se encuentra implementado en Haploview, se identifica un bloque haplotípico que comprende todos los SNP analizados excepto rs2516837. Adicionalmente el análisis de tag SNP muestra que para captar el 100% de la variabilidad genética observada en USF1 para los 9 marcadores seleccionados en la muestra poblacional analizada es necesaria sólo una combinación determinada de 6 de ellos debido a la estructura existente a nivel genético, que no era conocida a priori.
, y a R2, correspondiente al coeficiente de correlación entre alelos de cada SNP (B). Las cifras en los recuadros son los valores de los parámetros multiplicados por 100. Los recuadros sin cifra indican que el valor es máximo (1).")
Representación gráfica del desequilibrio de unión entre los SNP estudiados. Se muestran los diagramas elaborados con Haploview para el parámetro D’, correspondiente al desequilibrio de unión normalizado (A), y a R2, correspondiente al coeficiente de correlación entre alelos de cada SNP (B). Las cifras en los recuadros son los valores de los parámetros multiplicados por 100. Los recuadros sin cifra indican que el valor es máximo (1).
No se observó una asociación estadísticamente significativa con la condición de SM para ninguno de los polimorfismos estudiados en USF1 considerados aisladamente, tanto a nivel de frecuencias alélicas como de frecuencias genotípicas, tal como se muestra en la tabla 3.
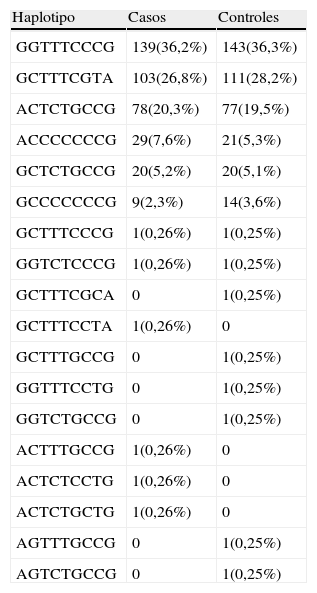
Se estimaron las frecuencias haplotípicas, y los haplotipos para cada muestra fueron inferidos mediante el algoritmo de Stephens23, que está implementado en el programa PHASE 2.1. Se detectaron un total de 18 haplotipos diferentes, como muestra la tabla 4, de los cuales 14 se encontraron en casos y 12 en controles, siendo 8 comunes a ambos grupos. El 98,2% de los alelos detectados en casos y controles corresponden a alguno de los 6 haplotipos más frecuentes, y el 97,2% de los sujetos estudiados son portadores de alelos de USF1 con alguno de estos haplotipos mayoritarios. El estudio de asociación de haplotipos de USF1 con SM no resultó estadísticamente significativo.
Frecuencias de los haplotipos estimados
| Haplotipo | Casos | Controles |
| GGTTTCCCG | 139(36,2%) | 143(36,3%) |
| GCTTTCGTA | 103(26,8%) | 111(28,2%) |
| ACTCTGCCG | 78(20,3%) | 77(19,5%) |
| ACCCCCCCG | 29(7,6%) | 21(5,3%) |
| GCTCTGCCG | 20(5,2%) | 20(5,1%) |
| GCCCCCCCG | 9(2,3%) | 14(3,6%) |
| GCTTTCCCG | 1(0,26%) | 1(0,25%) |
| GGTCTCCCG | 1(0,26%) | 1(0,25%) |
| GCTTTCGCA | 0 | 1(0,25%) |
| GCTTTCCTA | 1(0,26%) | 0 |
| GCTTTGCCG | 0 | 1(0,25%) |
| GGTTTCCTG | 0 | 1(0,25%) |
| GGTCTGCCG | 0 | 1(0,25%) |
| ACTTTGCCG | 1(0,26%) | 0 |
| ACTCTCCTG | 1(0,26%) | 0 |
| ACTCTGCTG | 1(0,26%) | 0 |
| AGTTTGCCG | 0 | 1(0,25%) |
| AGTCTGCCG | 0 | 1(0,25%) |
Siguiendo una metodología similar, se restringió el análisis de haplotipos a los polimorfismos que Pajukanta et al8 describieron como asociados a HLFC e hipertrigliceridemia, pero no se encontraron diferencias significativas que indicaran una asociación de estos haplotipos reducidos con el desarrollo de SM.
DiscusiónEl SM está adquiriendo proporciones cada vez mayores a nivel poblacional y constituye un importante problema clínico porque se asocia al desarrollo de enfermedad cardiovascular y diabetes tipo 2. Aunque su definición como entidad nosológica es controvertida, su utilidad a nivel clínico es indiscutible y existen evidencias que indicarían un componente genético en su manifestación.
Se ha descrito una heredabilidad del 24% para el SM, así como de sus diferentes componentes27, lo cual proporciona una base para la búsqueda de loci genéticos que confieran susceptibilidad para el desarrollo de SM. Se ha encontrado asociación de distintos genes relacionados con cada uno de los componentes individuales del SM con el desarrollo del propio SM, aunque no han podido ser reproducidos en todos los casos28. Estos resultados no concluyentes son posiblemente debidos al carácter genéticamente complejo que cabe atribuir al SM, en cuyo desarrollo pueden participar varios loci con variantes comunes, con la confusión que añade la influencia de factores ambientales que pueden afectar al fenotipo presentado, como son los niveles de ingesta calórica y actividad física, así como la importancia del género y la edad para su establecimiento. La definición de un fenotipo inequívoco para el SM también contribuye a la dificultad para identificar loci genéticos relacionados.
El solapamiento de rasgos fenotípicos del SM con determinados aspectos de la HLFC, y la identificación de USF1 como un gen causal para esta última, han hecho que sea considerado como un posible gen asociado al desarrollo de SM por diferentes grupos29. La asociación de USF1 con HLFC ha sido estudiada y replicada en distintas poblaciones30, pero los resultados relativos a SM o patologías relacionadas como la diabetes tipo 2 son escasos y contradictorios31–35.
Esta relación hipotética no ha sido evaluada previamente en población española, y nuestro estudio explora la variabilidad genética en USF1 en una muestra extraída de nuestro entorno y su posible relación con SM en un estudio caso-control. Para ello se han analizado 9 posiciones polimórficas distribuidas en USF1 con el fin de captar dicha variabilidad. Esto ha permitido definir 6 haplotipos en el locus USF1 comunes que están presentes en la mayoría de la población.
Los resultados obtenidos en el estudio caso-control realizado no apoyan la hipótesis de que USF1 pueda estar asociado en el desarrollo de SM. La distribución de frecuencias de los SNP, considerados aisladamente o formando parte de los haplotipos identificados, no difiere significativamente entre casos y controles como cabría esperar si la variabilidad en el locus USF1 tuviera una influencia en el desarrollo de SM. Tampoco se ha encontrado relación de los SNP identificados por Pajukanta et al8 en relación con HLFC.
Considerando que el SM puede estar causado por la contribución de diferentes genes, la interpretación más directa de nuestros resultados sería que la variabilidad genética en USF1 en población de nuestro entorno no está asociada particularmente con SM debido a las diferencias étnicas existentes respecto a otras poblaciones en las que sí se ha observado una asociación significativa, y que serían otros genes los que tuvieran mayor contribución al desarrollo de SM en nuestra población. Adicionalmente pueden existir interacciones entre genes causantes de SM, y USF1 no debería ser descartado como un gen con un efecto modificador de la acción de estos genes, como sugieren trabajos recientes que describen una interacción entre USF1 y APOA536.
Nuestro estudio tiene limitaciones que han podido influir parcialmente en el resultado obtenido. El tamaño muestral es adecuado para conseguir una potencia estadística razonable, pero si el efecto de USF1 fuera más débil de lo descrito en otras poblaciones para la población de nuestro entorno, el número de individuos analizados podría no haber sido suficiente para detectarlo. Adicionalmente los grupos de casos y controles no están perfectamente equilibrados en cuanto a la distribución de género y edad, y este hecho podría tener un efecto sobre el estudio de asociación para una situación fisiopatológica en la que edad y sexo tienen una gran influencia. Sin embargo el desequilibrio entre casos y controles en nuestro estudio es tal que el sesgo que podría haber sido introducido favorecería más bien una asociación positiva, dado que en el grupo de casos la edad es mayor y la proporción de hombres es mayor que la de mujeres, factores ambos que se asocian con el desarrollo de SM.
En conclusión, el estudio caso-control desarrollado en el presente trabajo no ha mostrado una asociación significativa de USF1 con el desarrollo de SM, lo cual refutaría su participación en el SM para la población de nuestro entorno, aunque todavía sería plausible su implicación como gen modificador de la acción de otros genes con mayor peso en el desarrollo de SM. La verificación de esta posibilidad requerirá el planteamiento de nuevos estudios que podrán tener en cuenta los resultados descritos aquí.
Contribución de los firmantes a la autoría de este trabajoDiseño del estudio: ALGO, FCM; Recogida y procesamiento muestras de pacientes: EJS, FCM, JPF, AMBS, ALGO; Obtención de datos: MS, JPF; Análisis e interpretación de datos: ALGO, MS; Redacción del borrador del artículo: ALGO; Revisión crítica del contenido del artículo: Todos; Aprobación final de la versión a publicar: Todos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Este trabajo ha sido realizado con la financiación de la beca FEA/SEA-Bristol Myers Squibb 2005. A.L.G.O. recibe apoyo del ISCIII/MICINN (PS09/00355) e I+CS (PIPAMER09/08).