




El factor tisular (FT) es una glucoproteína de la membrana iniciadora de la coagulación sanguínea. El contenido en FT predice el grado de trombogenicidad de la placa aterosclerótica1-4. Cuando la placa aterosclerótica se rompe, el lípido es un sustrato altamente trombogénico. La mayoría de los lípidos que se acumulan en la íntima arterial se encuentran en forma de lipoproteína de baja densidad (LDL) modificada por oxidación (LDLox) o por agregación (LDLag), procesos facilitados por la interacción de la LDL con las proteínas de la matriz extracelular que conforman la íntima arterial5,6. Tanto las LDL nativas como diferentes tipos de LDL modificadas incrementan la expresión de FT en diferentes tipos celulares. Sin embargo, solamente las LDLox y las LDLag tienen la capacidad de incrementar la actividad celular procoagulante7-9. La activación del FT por las LDLox se produce mediante peroxidación lipídica intracelular7,8. Nuestro grupo ha demostrado previamente que las LDLag se unen al receptor lipoproteico LRP1 (Low Density Lipoprotein Receptor-Related Protein). Este receptor media la captación selectiva y la acumulación intracelular de colesterol esterificado (CE) de la LDLag10-14. La captación de LDLag induce la expresión y la activación de FT y, por tanto, la transformación protrombótica de las células musculares lisas de la pared vascular (CMLV)9,14. Sabemos que la captación de LDLag por las CMLV no induce apoptosis, determinante crucial de desarrollo de aterotrombosis15,16. Sin embargo, desconocemos el mecanismo involucrado en la inducción FT por las LDLag. Diversos estudios han demostrado que los inhibidores de la HMG-CoA reductasa o estatinas son capaces de disminuir la incidencia de episodios coronarios gracias a sus efectos antitrombóticos17-19. Estos efectos beneficiosos incluyen la estabilización o regresión de la placa aterosclerótica. Las estatinas también han demostrado disminuir la expresión de FT en la aorta y también en las placas ateroscleróticas ricas en macrófagos presentes en conejos hipercolesterolémicos20-22. Nuestro grupo ha demostrado anteriormente que las estatinas inhiben significativamente la formación del trombo tanto en la pared vascular erosionada como en la dañada en modelo porcino23. Estos estudios in vivo demuestran una reducción en la trombogenicidad y una disminución en la expresión de FT en la pared vascular, y estos efectos son independientes de la disminución del colesterol sérico causada por las estatinas (efectos pleiotrópicos)20-23. Los efectos pleiotrópicos de las estatinas son el resultado de la capacidad de estos inhibidores para bloquear la síntesis de los principales donadores de isoprenoides hidrofóbicos, como son el farnesil pirofosfato (FPP) y el geranilgeranil pirofosfato (GGPP). Estos grupos son responsables de la modificación lipídica postraduccional (prenilación) de proteínas isopreniladas envueltas en señal intracelular, tales como las proteínas GTPasa pequeñas pertenecientes a las familias Rho y Ras24,25. El objetivo de este trabajo fue investigar el mecanismo involucrado en la inducción de FT por LDLag y la regulación de dicho mecanismo por la pravastatina.
Nuestros resultados demuestran que la pravastatina previene la inducción de FT provocada por la captación de LDLag y que este efecto se revierte en presencia de GGPP pero no de FPP, lo que sugiere que en la inducción de FT por LDLag interviene una proteína geranilgeranilada. De hecho, la captación de LDLag induce la translocación de Rho A del citoplasma (Rho A inactivo) a la membrana (Rho A activo), y la exoenzima C3, un inhibidor específico de Rho A, previene la activación de FT inducida por la LDLag. Estos resultados evidencian que la LDLag induce la expresión y la activación de FT, al menos en parte, mediante la tranlocación de Rho A a la membrana, y que la pravastatina, al inhibir la modificación postrasduccional de esta proteína, previene la activación de FT por captación de LDLag. Éste podría ser un nuevo mecanismo por el que la pravastatina previene la transformación protrombótica de las lesiones ateroscleróicas avanzadas.
Material y métodos
Cultivo de CMLV y preparación de las LDL
Los cultivos primarios de CMLV fueron obtenidos por la técnica de explantes a partir de zonas no ateroscleróticas de coronarias humanas aisladas de corazones trasplantados en el Hospital de la Santa Creu i Sant Pau10-12. El estudio fue aprobado por el Comité Revisor Institucional de Investigación Humana del Hospital de la Santa Creu i Sant Pau en concordancia con la Declaración de Helsinki. Las células quiescentes se preincubaron durante 4 h con un inhibidor de la HMG-CoA reductasa (pravastatina, cedida amablemente por los laboratorios Bristol-Myers Squibb), con pravastatina y con los diferentes intermediarios de la vía de síntesis endógena de colesterol, mevalonato (Sigma) (100 mM); GGPP (Sigma) (10 mM), FPP (Sigma) (10 mM) que se mantuvieron durante el tratamiento con LDL nativa (LDLn) o LDLag (100 mg/ml, 18 h). Para estudiar el efecto de la inhibición de Rho A, las CMLV quiescentes se preincubaron con exoenzima C3 (25 mg/ml, 24 h), inhibidor que se mantuvo durante el período de la incubación con lipoproteínas (18 h). Los inhibidores o productos de la vía de síntesis de colesterol no influyeron en la viabilidad de las células (ensayos realizados por tinción con azul de tripano).
El aislamiento de las LDL humanas (d1.019-d1.063 g/ml) se realizó mediante ultracentrifugación secuencial a partir de una mezcla de sueros normocolesterolémicos de voluntarios. Las LDLag se prepararon por agitación mecánica de la LDL, tal como se describió anteriormente9-14. Los valores de TBARS (< 1,2 mmoles de malondialdehído/mg proteína en LDL) se mantuvieron similares a los observados en las LDLn después de la agregación.
PCR a tiempo real
El ARN total y la proteína celular fueron aislados utilizando Tripure Isolation Reagent (Roche Molecular Biochemicals) siguiendo las indicaciones del fabricante. Los valores de ARNm de FT se analizaron por PCR a tiempo real. Los cebadores y la sonda fluorescente TaqMan de FT fueron diseñados utilizando el programa Primer Express (PE Systems)9. Se utilizó GAPDH humana como gen endógeno (4326317E Applied Biosystems). La PCR a tiempo real TaqMan se realizó como se ha descrito anteriormente9.
Determinación de la actividad procoagulante de FT en CMLV
La actividad procoagulante de FT (APC) se testó por un método de generación de factor X activado (Xa) con mínimas modificaciones26,27. Se utilizó un sustrato cromogénico de factor Xa (Sigma) (0,5 mM) y la actividad procoagulante se determinó por colorimetría midiendo la densidad óptica a 405 nm utilizando un lector de ELISA a 37 °C (SpectraMax). Los valores se obtuvieron a partir de una curva estándar realizada con factor Xa (Calbiochem).
Translocación de Rho A del citoplasma a la membrana celular
Las CMLV humanas se expusieron a LDL después de haber sido incubadas en ausencia o presencia de pravastatina (0,5 mM). Se obtuvieron las fracciones proteicas del citoplasma y la membrana utilizando ProteoExtract® Subcellular Proteome Extraction kit (Calbiochem), que se purificaron mediante el kit 2D Clean-Up (Amersham). El análisis de Rho A en el citoplasma y la membrana se realizó mediante Western blot incubando con anticuerpos monoclonales contra Rho A humana (Santa Cruz, 26C4).
Análisis de datos
Los datos se expresaron como la media ± SEM. Para el análisis de los datos se utilizó un paquete estadístico para Macintosh: statview (Abacus Concepts). Los grupos múltiples se analizaron por ANOVA o test de Wilcoxon, dependiendo de las necesidades. Se consideraron estadísticamente significativos los valores con p < 0,05.
Resultados
La pravastatina disminuye la sobreexpresión y la activación de FT inducidas por la captación de LDLag en CMLV humanas
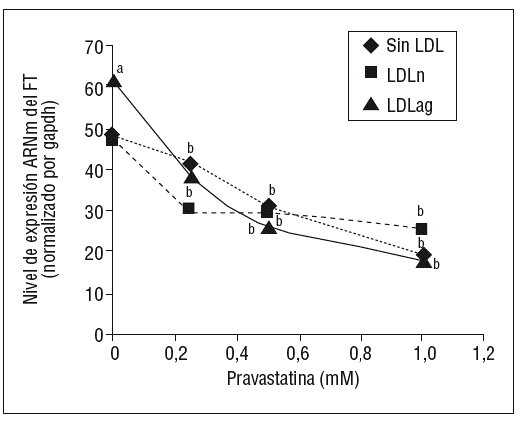
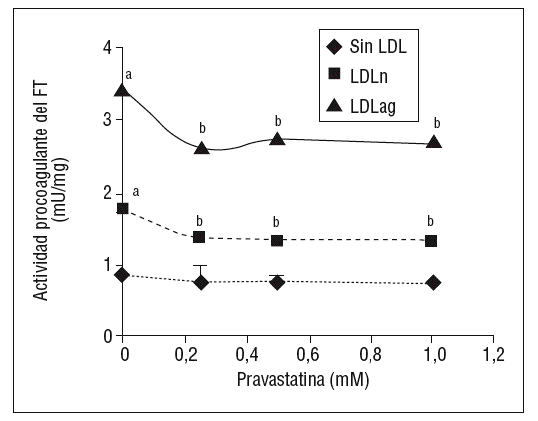
Como se muestra en la figura 1, la pravastatina (0,25 mM, 0,5 mM, 1 mM) disminuyó la expresión de ARNm de FT de forma dependiente de la dosis en CMLV. La disminución de los valores de ARNm de FT a dosis de 1 mM es del 59 ± 0,57% en CMLV control, del 47 ± 0,09% en CMLV expuestas a LDLn y del 70 ± 0,29% en CMLV expuestas a LDLag. La LDLag incrementó de forma muy marcada la APC de FT en CMLV (LDLag: 3,44 ± 0,21, LDLn: 1,75 ± 0,19 y control: 0,84 ± 0,04 U/mg proteína) en concordancia con resultados previos obtenidos en nuestro grupo9. Como se observa en la figura 2, la pravastatina disminuyó el incremento en APC inducido por LDLag desde el 24 ± 0,68% a dosis de 0,25 mM hasta el 39 ± 0,56% a dosis de 1 mM. La pravastatina también disminuyó significativamente la actividad de FT en CMLV expuestas a LDLn, pero no ejerció ningún efecto en las CMLV control.
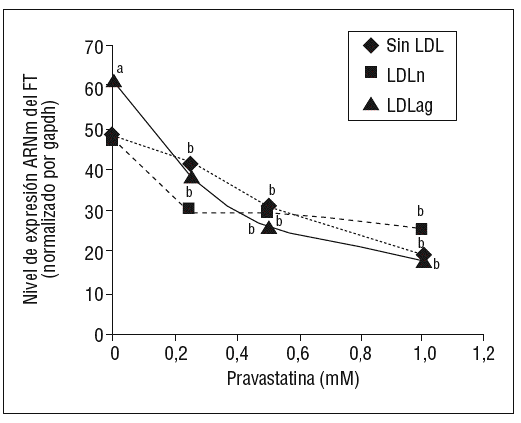
Figura 1. Efecto de la pravastatina sobre los niveles de expresión de ARNm de FT inducidos por LDLag. El gráfico muestra los niveles de expresión de ARNm de FT obtenidos a partir de la cuantificación por PCR a tiempo real. Las CMLV se preincubaron durante 4 h con pravastatina (0, 0,25, 0,5, 1 mM) y luego se incubaron durante 18 h sin LDL (CMLV controles, rombos), con LDLn (cuadrados) o con LDLag (triángulos) (100 mg/ml). Los datos se procesaron utilizando un programa especialmente diseñado y basado en los Ct de la muestra normalizados respecto al ARNm de gapdh (n = 3). p < 0,05: avs CMLV no expuestas a LDL; bvs CMLV sin tratar con pravastatina; CMLV: células musculares lisas de la pared vascular; FT: factor tisular; LDL: lipoproteína de baja densidad; LDLag: LDL modificada por agregación; LDLn: LDL nativa; PCR: reacción en cadena de la polimerasa.
El GGPP, pero no el FPP, revierte el efecto de la pravastatina en la expresión y la actividad de FT inducida por captación de LDLag
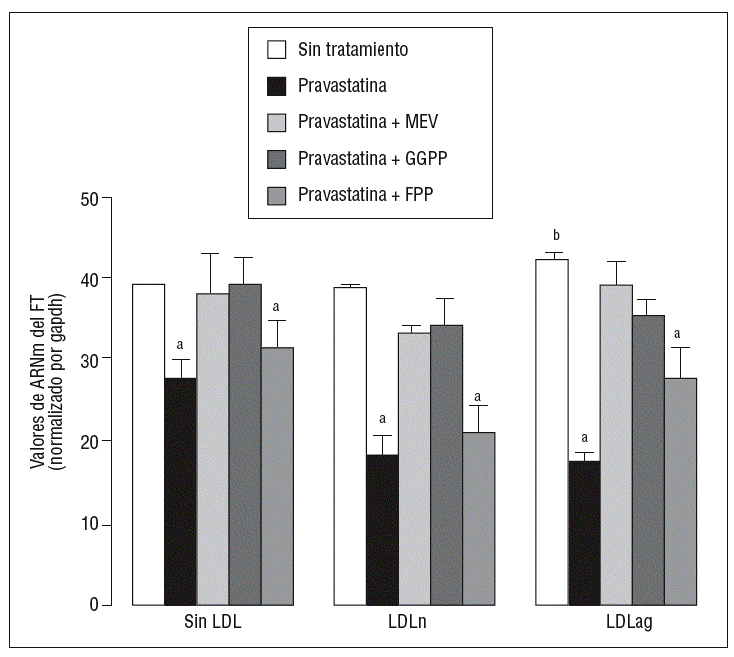
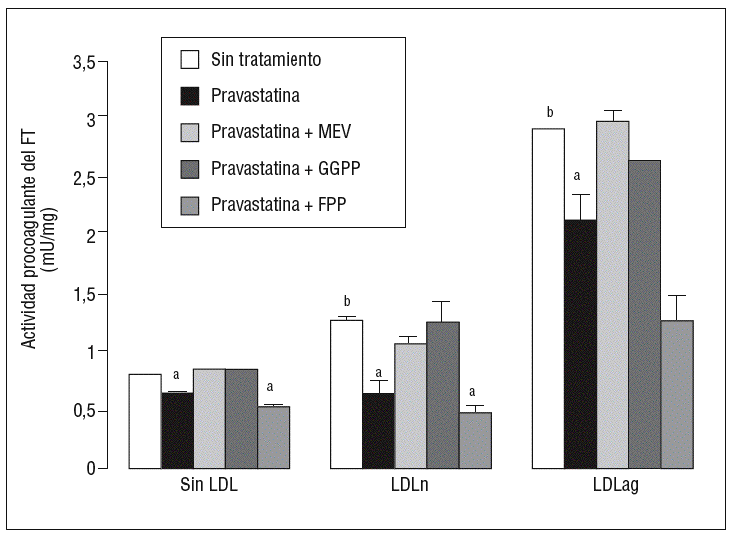
Para caracterizar los principales grupos isoprenilados involucrados en el efecto de la pravastatina adicionamos mevalonato, GGPP o FPP durante el tratamiento con este inhibidor. En la figura 3 se observa que el mevalonato y el GGPP revirtieron el efecto de la pravastatina en la expresión de ARNm de FT tanto en ausencia como en presencia de LDL. Por el contrario, el FPP no revirtió el efecto de la pravastatina en ningún caso. Como se muestra en la figura 4, el mevalonato y el GGPP también previnieron completamente el efecto de la pravastatina sobre la actividad procoagulante de FT en CMLV expuestas a LDLn o LDLag, mientras que el FPP no revirtió el efecto de la estatina en ninguna de las condiciones. Ninguno de los grupos isoprenil ejerció por sí mismo efecto sobre la actividad del FT. Estos resultados sugieren que en la inducción de la expresión y la activación de FT por LDLag interviene una proteína geranilgeranilada.
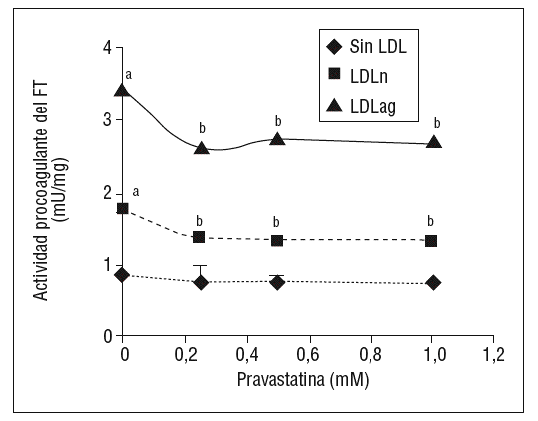
Figura 2. Efecto de la pravastatina sobre la actividad procoagulante del FT inducida por LDLag. El gráfico muestra la actividad medida a través de un método colorimétrico de generación de factor Xa. Las CMLV se preincubaron durante 4 h con pravastatina (0, 0,25, 0,5, 1 mM) y luego se incubaron durante 18 h sin LDL (CMLV controles, rombos), con LDLn (cuadrados) o con LDLag (triángulos) (100 mg/ml). Los re sultados se expresan en U/mg prot y se muestran como media ± SEM (de 3 experimentos con triplicados). p < 0,05: avs CMLV sin incubar con LDL; bvs CMLV sin tratar con pravastatina; CMLV: células musculares lisas de la pared vascular; FT: factor tisular; LDL: lipoproteína de baja densidad; LDLag; LDL modificada por agregación; LDLn: LDL nativa.
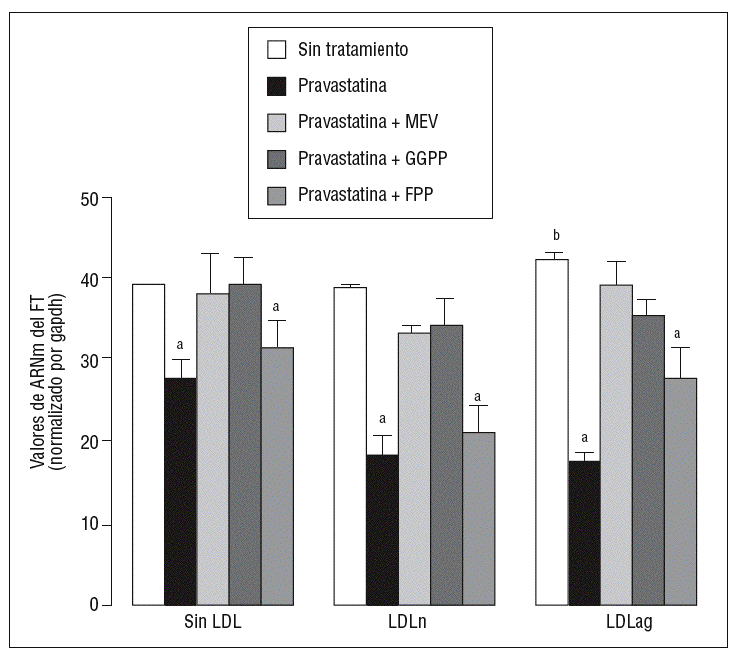
Figura 3. Reversión por GGPP del efecto de la pravastatina sobre los valores de ARNm del FT inducidos por LDLag. El gráfico muestra los niveles de expresión de ARNm de FT obtenidos a partir de la cuantificación por PCR a tiempo real. Las CMLV se preincubaron durante 4 h con pravastatina (0,5 mM) y en ausencia o presencia de mevalonato (0,1 mM), GGPP (10 mM) o FPP (10 mM). Posteriormente, las CMLV se incubaron durante 18 h sin LDL, con LDLn o con LDLag (100 mg/ml). Los datos se procesaron utilizando un programa especialmente diseñado y basado en los Ct de la muestra normalizados respecto al ARNm de gapdh (n = 3). p < 0,05: avs CMLV sin tratar con pravastatina; bvs CMLV sin incubar con LDL; CMLV: células musculares lisas de la pared vascular; FPP: farnesil pirofosfato; FT: factor tisular; GGPP: geranilgeranil pirofosfato; LDL: lipoproteína de baja densidad; LDLag: LDL modificada por agregación; LDLn: LDL nativa; MEV: mevalonato.
La LDLag induce la translocación de Rho A del citoplasma a la membrana celular y la pravastatina inhibe este efecto
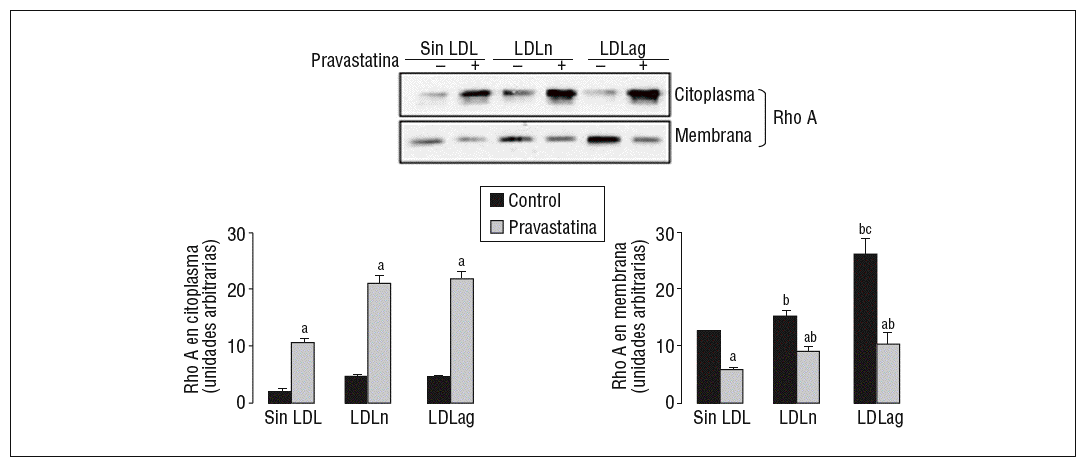
Como se muestra en la figura 5, la LDLag incrementó el valor proteico de Rho A en la membrana (forma activa) (agLDL: 2,10 veces frente a nLDL: 1,21 veces). Tanto la LDLag como la LDLn indujeron un ligero incremento en la forma citoplasmática (inactiva) de Rho A. La captación de LDLag no ejerció ningún efecto significativo en los valores de Rac1 en el citoplasma o la membrana (datos no mostrados). El aumento de la forma activa de Rho A inducido por LDLag se asocia con un incremento en la expresión de ARNm de Rho A (agLDL: 2 veces frente a LDLn: 1,22 veces). La pravastatina redujo significativamente la forma activa de Rho A en CMLV control (en un 54 ± 0,2%), en CMLV expuestas a LDLn (en un 39 ± 0,5%) y en CMLV expuestas a LDLag (en un 61 ± 2,2%). La pravastatina indujo un incremento en la forma citoplasmática (inactiva de Rho A) tanto en CMLV control (5,48 ± 0,5 veces) como en CMLV expuestas a LDLn (4,74 ± 1,5 veces) como en CMLV expuestas a LDLag (9,68 ± 1,4 veces).
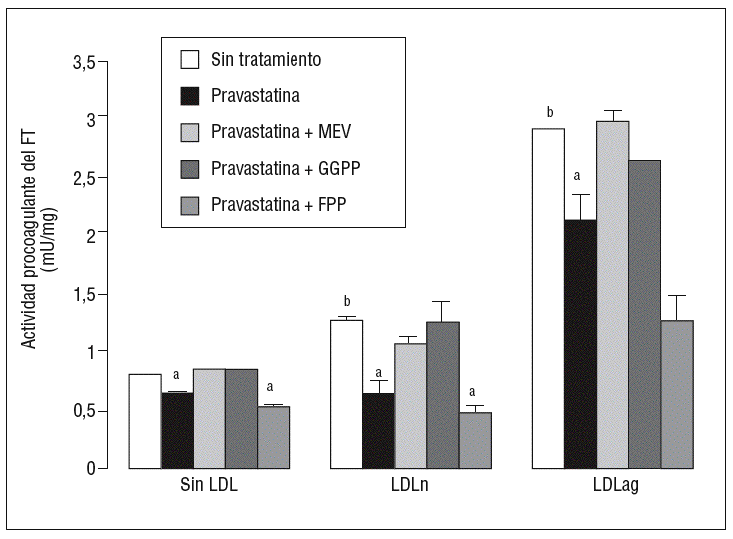
Figura 4. Reversión por GGPP del efecto de la pravastatina sobre la actividad procoagulante del FT inducida por LDLag. El gráfico muestra la actividad medida a través de un método colorimétrico de generación de factor Xa. Las CMLV se preincubaron durante 4 h con pravastatina (0,5 mM) y en ausencia o presencia de mevalonato (0,1 mM), GGPP (10 mM) o FPP (10 mM). Posteriormente, se incubaron durante 18 h sin LDL, con LDLn o con LDLag (100 mg/ml). Los resultados se expresan en U/mg prot y se muestran como media ± SEM (de 3 experimentos con triplicados). p < 0,05: avs CMLV sin tratar con pravastatina; bvs CMLV sin incubar con LDL; CMLV: células musculares lisas de la pared vascular; FPP: farnesil pirofosfato; FT: factor tisular; GGPP: geranilgeranil pirofosfato; LDL: lipoproteína de baja densidad; LDLag: LDL modificada por agregación; LDLn: LDL nativa; MEV: mevalonato.
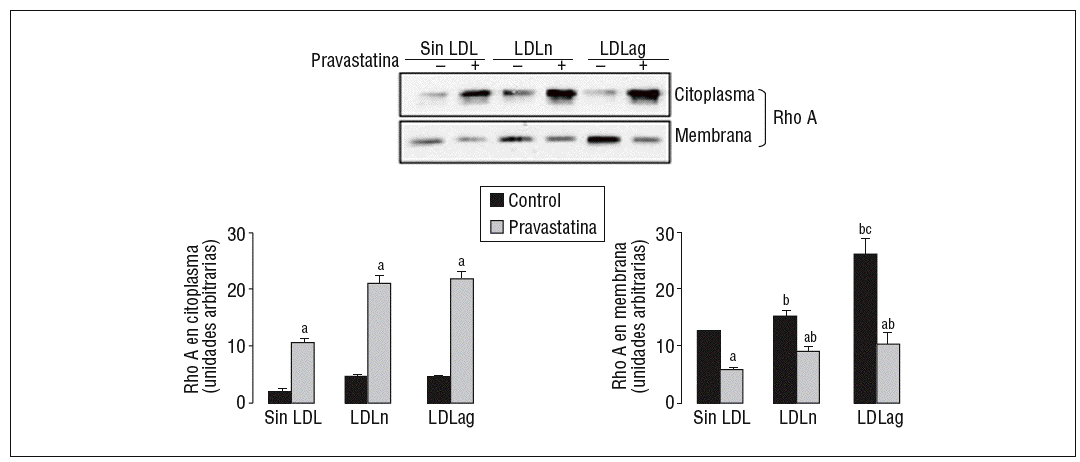
Figura 5. Efecto de la LDLag en la translocación de Rho A del citoplasma a la membrana. Las CMLV control y las CMLV tratadas con pravastatina (0,5 mM, 4 h) se incubaron posteriormente durante 18 h sin LDL, con LDLn o con LDLag (100 mg/ml). Se muestra un Western blot representativo de la proteína Rho A presente en la fracción citoplasmática (inactiva) y la fracción de la membrana (activa), así como un diagrama de barras con la cuantificación de bandas. Los resultados se expresan como unidades arbitrarias y se muestran como la media ± SEM (n = 3). p < 0,05: avs CMLV sin tratar con pravastatina; bvs CMLV control; cvs CMLV expuestas a LDLn. CMLV: células musculares lisas de la pared celular; LDL: lipoproteínas de baja densidad; LDLag: modificada por agresión; LDLn: LDL negativa.
La inhibición específica de Rho A previene la inducción de la expresión y la activación de FT por LDLag
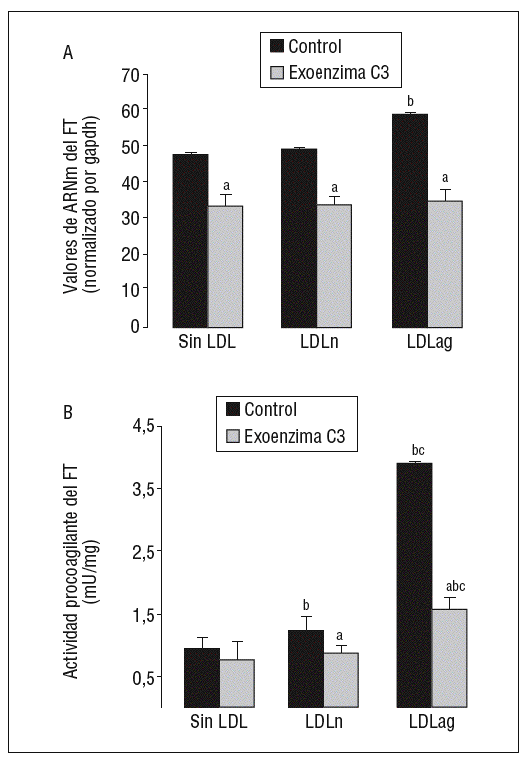
Como se observa en la figura 6, la exoenzima C3, un inhibidor específico de Rho A (25 mg/ml, 24 h), inhibió la expresión basal de ARNm de FT en un 31 ± 3% así como la inducción de la expresión de ARNm de FT inducida por LDLn (en el 30 ± 2,1%) o LDLag (en el 42 ± 3,3%) (fig. 6A). La exoenzima C3 previno tanto la ligera activación de FT inducida por LDLn como la fuerte activación de FT inducida por la LDLag en el 41,4 ± 0,14% (fig. 6B).
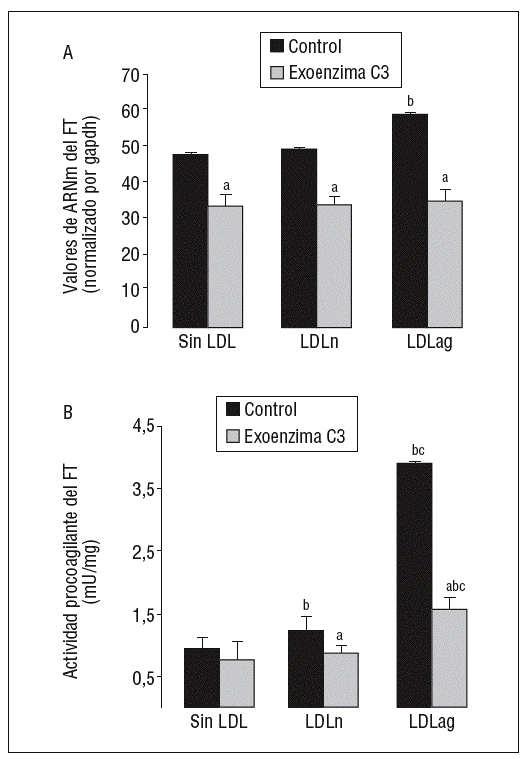
Figura 6. La exoenzima C3 (un inhibidor de Rho A) previene la expresión y la activación de FT inducida por LDLag. Las CMLV control y las CMLV tratadas con exoenzima C3 (25 mg/ml, 24 h) se incubaron posteriormente durante 18 h sin LDL, con LDLn o con LDLag (100 mg/ml). A) El gráfico muestra los niveles de expresión de ARNm de FT obtenidos a partir de la cuantificación por PCR a tiempo real. Los datos se procesaron utilizando un programa especialmente diseñado y basado en los Ct de la muestra normalizados respecto al ARNm de gapdh (n = 3). B) El gráfico muestra la actividad procoagulante de FT. Los resultados se expresan en U/mg prot y se muestran como media ± SEM (de 3 experimentos con triplicados). p < 0,05: avs CMLV sin tratar con pravastatina; bvs CMLV control; cvs CMLV expuestas a LDLn. CMLV: células musculares lisas de la pared celular; FT: factor tisular; LDL: lipoproteínas de baja densidad; LDLag: LDL modificada por agregación; LDLn: LDL nativa.
Discusión
Las CMLV son una de las principales fuentes de FT en la pared vascular1-4. Teniendo en cuenta la importancia de las placas ateroscleróticas erosionadas, ricas en CMLV, en el desencadenamiento de la trombosis28,29, los mecanismos por los que se induce el FT en CMLV han ganado interés. Diversos estudios han evidenciado que las estatinas influyen en la expresión y la actividad de FT en varios modelos, tanto in vivo como in vitro19-23. Aunque sabemos que la LDLag, una de las principales modificaciones de la LDL en la íntima arterial, con alta capacidad de provocar la formación de células espumosas10-14, induce la expresión y la activación del FT en CMLV humanas, sin inducir apoptosis12, desconocemos qué intermediarios celulares están involucrados en la inducción de FT por LDLag.
Nuestros resultados demuestran que la pravastatina previene la inducción del FT por LDLag tanto en expresión como en grado de activación. Sin embargo, se observa una eficacia diferencial de la pravastatina en la inhibición de los efectos de la LDLag sobre la expresión de ARNm de FT y la actividad procoagulante de FT. Estos resultados sugieren que mientras que la LDLag induce la sobreexpresión de FT por un mecanismo inhibible por estatinas, la LDLag podría inducir la actividad procoagulante de FT por otros mecanismos no inhibibles por estatinas. Estudios previos sugieren que la expresión y la activación de FT son procesos independientes30,31. La activación de FT está principalmente regulada por mecanismos que alteran su estructura cuaternaria32. Además, en nuestro grupo hemos demostrado recientemente que la LDLag tiene la capacidad de alterar la concentración de esfingomielina (SM) de la membrana plasmática de las CMLV9. La SM es uno de los principales fosfolípidos que estructuran las caveolas, estructuras altamente enriquecidas en FT en su forma encriptada33. Por lo tanto, al alterar el contenido de SM de la membrana plasmática, la LDLag podría influir en la topología y, por consiguiente, en la activación de FT. El efecto de la pravastatina sobre los valores de ARNm y la actividad de FT inducidos por la captación de LDLag se revierte por GGPP, pero no por FPP, lo que sugiere la implicación de una proteína geranilgeranilada en la inducción de FT por LDLag. En este estudio demostramos que la LDLag tiene una capacidad muy superior a la LDLn para inducir la translocación de Rho A desde el citoplasma a la membrana celular, donde está activa. La pravastatina disminuyó en mayor grado la forma activa de Rho A en CMLV tratadas con LDLag o controles que en CMLV tratadas con LDLn. Ello podría ser debido a que los inhibidores de la HMG-CoA reductasa inducen la expresión del receptor clásico de LDL favoreciendo una sobrecaptación de LDL y, por tanto, una sobreinducción de Rho A activo que compensaría en cierta medida la inhibición de la síntesis de GGPP inducida por la pravastatina en CMLV expuestas a LDLn. La pravastatina inhibe la síntesis de GGPP necesaria para la modificación postraduccional de Rho A, su translocación a la membrana y, por tanto, su activación. Un inhibidor específico de Rho A, la exoenzima C3, disminuye la inducción por LDLag de los niveles de expresión de FT y la inducción de la activación de FT por las LDLag prácticamente hasta el nivel basal. Conjuntamente estos resultados demuestran que la inducción de FT por la LDLag depende de la translocación a la membrana de la proteína Rho A, y que la pravastatina previene la inducción de FT inducida por LDLag al bloquear la síntesis del GGPP necesaria para el anclaje a la membrana de Rho A. Nuestros resultados concuerdan con estudios previos que demuestran que las estatinas impiden la translocación a la membrana de Rho A, influyendo así en la expresión de FT en monocitos y células endoteliales34,35. Lo novedoso de este estudio es que demostramos por primera vez que las LDLag inducen la translocación de Rho A de forma significativa, dando una explicación mecanística a la alta capacidad protrombótica de esta lipoproteína modificada9,14. Diferentes agonistas, como las LDL oxidadas, la trombina y la angiotensina II, inducen el FT a través de la activación de la vía Rho/Rho cinasa en diferentes modelos celulares34,36,37, indicando que la activación de Rho A está involucrada en la activación del FT por varios agonistas que se unen a diferentes receptores sin relación entre ellos. La potencial importancia de la vía de Rho/Rho cinasa en la aterogénesis ha sido demostrada en modelos animales, en los que tratamientos con inhibidores de Rho cinasa redujeron el desarrollo de la lesión38. Aunque la LDLn también induce Rho A, la captación de LDLn está limitada por la regulación a la baja de su LRP1 es un proceso no regulado por colesterol que permite una acumulación muy elevada de colesterol intracelular9-14. Ello es debido a que el LRP1 se regula al alza por LDLag12 y a que el LRP1 participa en un mecanismo de captación selectiva de CE13. Nuestro estudio contribuye a evidenciar el importante papel atribuido a Rho A en la patogénesis de la arteriosclerosis38, ya que la captación de LDLag mediada por LRP1 podría ser un mecanismo proaterotrombótico de gran relevancia en la pared vascular14.
Agradecimientos
Los autores agradecen la colaboración del Equipo de Trasplantes del Servicio de Cardiología y Enfermedad Cardíaca del Hospital de la Santa Creu y Sant Pau y al Banco de Sangre del Hospital Clínic de Barcelona. También dan las gracias a Laura Nasarre y a Vanessa Martín por su soporte técnico.