




En esta revisión se valora la contribución del tejido adiposo blanco en las complicaciones vasculares asociadas a la obesidad. El tejido adiposo blanco es un órgano metabólicamente activo y secretor de distintas moléculas que tienen una acción endocrina, paracrina y autocrina. El aumento de peso producido en la obesidad genera un exceso de grasa, generalmente visceral, responsable de la puesta en marcha de vías de señalización que activan la producción de citoquinas y quimioquinas proinflamatorias. Tanto los adipocitos como los macrófagos y linfocitos infiltrados y las células endoteliales contribuyen a la situación crónica inflamatoria de bajo grado presente en la obesidad. Además, el aumento de adiposidad no solo activa la respuesta inflamatoria en el propio adipocito, sino también en el hepatocito. Finalmente, los mediadores proinflamatorios y proaterogénicos que son producidos por el tejido adiposo blanco y el hígado y asociados a las células inmunes generan una inflamación sistémica que produce resistencia a la insulina en tejidos periféricos, además de contribuir al inicio del proceso aterogénico.
The contribution of white adipose tissue to the vascular complications associated with obesity is analysed in this review. White adipose tissue is an active metabolic organ and secretor of several molecules with endocrine, paracrine and autocrine actions. Weight gain produced in the obesity, induces an excess of fat, mainly in the visceral depot, which is responsible for the activation of different signalling pathways, leading to a higher production of proinflammatory cytokines. As adipocytes as infiltrated macrophages and lymphocytes and endothelial cells contribute to a chronic low grade inflammatory situation present in obesity. Moreover, the increase in adiposity activates the inflammatory response in the adipocyte themselves, as well as in the hepatocyte. Finally, proinflammatory and proatherogenic mediators produced by white adipose tissue and liver associated to immune cells generate insulin resistance in peripheral tissues and contribute to the beginning of atherogenic process.
En las últimas décadas, la obesidad ha pasado de ser un problema estético a ser una verdadera epidemia que afecta a más de un tercio de la población occidental, y que comienza a afectar cada día más a las generaciones jóvenes1,2.
La obesidad es una enfermedad crónica de origen multifactorial que tiene un riesgo cardiovascular elevado por la coexistencia de otros factores de riesgo, como la dislipidemia, la hipertensión, la resistencia a la insulina y la diabetes3,4. De hecho, estos factores de riesgo están íntimamente ligados a un exceso de tejido adiposo, y más específicamente a su particular distribución corporal. Esta forma de distribución de la grasa en el obeso sí está claramente relacionada de manera independiente con la morbimortalidad cardiovascular a través de un síndrome metabólico aterogénico5. Este es el motivo por el que adquiere especial trascendencia clínica la medida, no solo de la cuantía total de la grasa corporal, sino de su distribución, habida cuenta que tal distribución tiene más impacto en el riesgo cardiovascular que la obesidad por sí misma6.
En los humanos hay 2 tipos de tejidos adiposos: el tejido adiposo blanco (WAT) y el tejido adiposo marrón o pardo (BAT). Ambos tejidos adiposos presentan diferencias importantes con relación a su funcionalidad, su morfología y su distribución. El WAT, además de ser el principal reservorio de energía, es un órgano secretor de un gran número de hormonas y citoquinas que modulan el metabolismo del organismo5-8. Por otro lado, el BAT está especializado en el gasto energético a través de la termogénesis. Por tanto, el desarrollo de la obesidad depende del balance entre el WAT y el BAT. En esta revisión nos hemos centrado en analizar el papel del WAT en la obesidad, con especial interés en las alteraciones vasculares asociadas a ella, que por otro lado constituyen las principales causas de morbimortalidad en el mundo occidental.
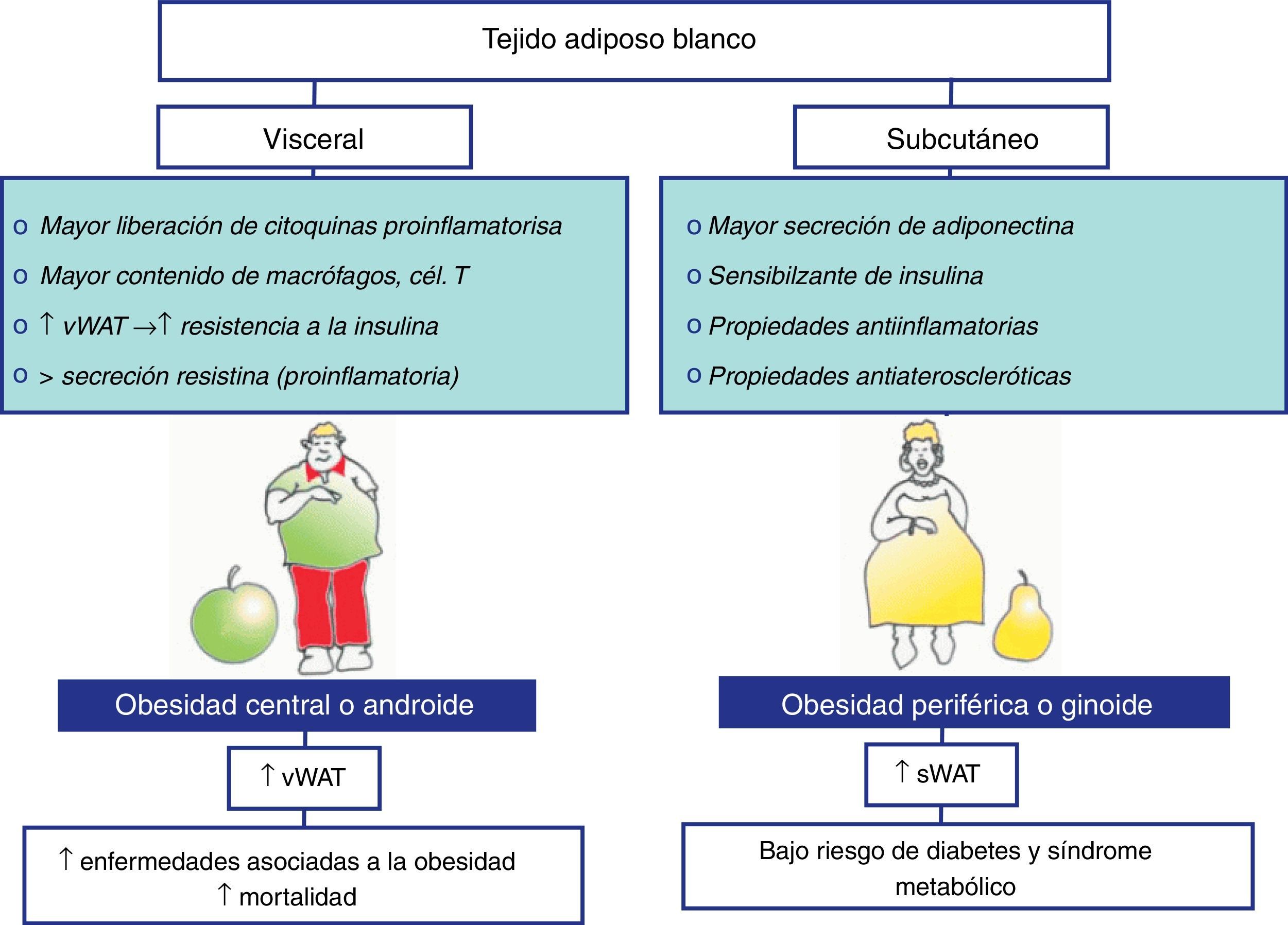
Papel del tejido adiposo blanco en la obesidad y sus complicaciones metabólicas y vasculares asociadasMorfología y distribución del tejido adiposo blanco. Implicación en el riesgo cardiovascularEl adipocito del WAT tiene una forma variable, aunque clásicamente es esférica y con un tamaño de entre 25 y 200μm. Además, tiene un núcleo periférico y plano y un citoplasma delgado que contiene una única gota lipídica grande que ocupa el 90% del volumen. Presenta escasas mitocondrias y un pequeño retículo endoplasmático liso y rugoso. El WAT se compone de los adipocitos que se mantienen unidos por un tejido conectivo laxo que está adecuadamente vascularizado e inervado9. Además de los adipocitos, el WAT contiene macrófagos, leucocitos, fibroblastos, adipocitos, células progenitoras y células endoteliales. La presencia de los fibroblastos, los macrófagos y otros leucocitos, junto con los adipocitos, da cuenta de la gran variedad de proteínas que son secretadas por el WAT bajo condiciones variables. El WAT está distribuido a lo largo de todo el organismo y tiene diferentes compartimentos que varían en cuanto al tamaño celular del adipocito10,11, a la actividad metabólica y a su papel potencial en la resistencia a la insulina y otras complicaciones vasculares asociadas a la obesidad12,13. En los humanos se diferencian 2 depósitos principales de WAT: el depósito subcutáneo, correspondiente al tejido adiposo que se localiza bajo la piel, y el depósito visceral. Hay 2 tipos de tejido adiposo visceral: el mesentérico y el omental14. El primero se encuentra envolviendo al intestino, y el segundo se extiende desde la parte inferior del estómago, recubriendo el abdomen, y es el que normalmente se emplea en el estudio de la grasa visceral. Hace tiempo que se sabe que el tejido adiposo visceral y el subcutáneo presentan numerosas diferencias anatómicas, celulares y moleculares15,16; por ejemplo, la irrigación de ambos tejidos es diferente17, y los niveles de ARN mensajero de leptina en el tejido adiposo subcutáneo están incrementados respecto del visceral15. Estos tejidos también son diferentes en cuanto a la capacidad de movilización de ácidos grasos, la grasa omental es más sensible a los efectos lipolíticos de las catecolaminas y menos sensible a los efectos antilipolíticos de la insulina; por tanto, este tejido tiene una mayor capacidad de movilización de ácidos grasos que el depósito subcutáneo17,18. Adicionalmente se han descrito numerosas diferencias entre el tejido adiposo visceral y subcutáneo referentes a la secreción de adipoquinas19. En este sentido, una obesidad periférica se caracteriza por una acumulación de tejido adiposo subcutáneo y es más frecuente en mujeres. Este tipo de obesidad no se ha asociado a un mayor riesgo de sufrir patologías asociadas20. Sin embargo, la obesidad central o abdominal es más frecuente en hombres y consiste en una acumulación de tejido adiposo visceral. Este tipo de obesidad se ha asociado, mediante estudios epidemiológicos, con un mayor riesgo de padecer enfermedades tales como resistencia a la insulina, diabetes mellitus tipo2 e hipertensión, aumentando considerablemente el riesgo cardiovascular21 (fig. 1).
 visceral y subcutáneo. En ambos tejidos adiposos existen diferencias en cuanto a su capacidad de movilización de ácidos grasos, su sensibilidad a los efectos lipolíticos de las catecolaminas y antilipolíticos de la insulina, además de una diferencial secreción de adipoquinas. En este sentido, se ha descrito que la obesidad periférica, caracterizada por una acumulación del tejido subcutáneo, no se ha asociado a un mayor riesgo de sufrir patologías asociadas. Sin embargo, la obesidad central o abdominal, que es más frecuente en hombres, se caracteriza por una acumulación del tejido adiposo visceral y se asocia con un mayor riesgo de padecer enfermedades como la resistencia a la insulina o la hipertensión, aumentando considerablemente el riesgo cardiovascular.")
Diferencias entre el tejido adiposo blanco (WAT) visceral y subcutáneo. En ambos tejidos adiposos existen diferencias en cuanto a su capacidad de movilización de ácidos grasos, su sensibilidad a los efectos lipolíticos de las catecolaminas y antilipolíticos de la insulina, además de una diferencial secreción de adipoquinas. En este sentido, se ha descrito que la obesidad periférica, caracterizada por una acumulación del tejido subcutáneo, no se ha asociado a un mayor riesgo de sufrir patologías asociadas. Sin embargo, la obesidad central o abdominal, que es más frecuente en hombres, se caracteriza por una acumulación del tejido adiposo visceral y se asocia con un mayor riesgo de padecer enfermedades como la resistencia a la insulina o la hipertensión, aumentando considerablemente el riesgo cardiovascular.
En primer lugar, el WAT es un órgano que constituye el mayor reservorio energético del organismo. La energía es almacenada en los adipocitos en forma de triglicéridos. La principal fuente de triglicéridos procede de los quilomicrones y las proteínas de muy baja densidad (VLDL) circulantes. En los humanos, el almacenamiento de los ácidos grasos en el tejido adiposo depende prácticamente de su liberación desde las lipoproteínas por acción de la lipoproteína lipasa (LPL)22. Tal es el protagonismo de esta enzima en el metabolismo lipídico, que se describe una acción proaterogénica de la LPL, expresada por el macrófago, y una acción antiaterogénica de la LPL, expresada en el tejido adiposo y en el músculo. Por tanto, esta enzima estaría implicada en las alteraciones lipídicas de la obesidad23. Su actividad aumenta en el período posprandial y se inhibe en el ayuno, y está incrementada en el tejido adiposo tanto de humanos como de animales de experimentación obesos24-29. Sin embargo, la capacidad de respuesta de la LPL a la insulina y a la alimentación en pacientes obesos está disminuida27,30.
Otro de los procesos metabólicos que se producen en el tejido adiposo es la lipólisis, donde los triglicéridos almacenados en el tejido adiposo son hidrolizados a ácidos grasos y glicerol. El paso limitante de la lipólisis está controlado por la lipasa sensible a hormonas (HSL). Dicha hormona presenta una intensa regulación. Así, la activación de los receptores β-adrenérgicos produce un aumento de los niveles intracelulares de AMPc y estimula la fosforilación activadora (P-Ser 552), catalizada por la proteína quinasa activada por AMPc (PKA) de la HSL31. Sin embargo, la activación de los receptores α2-adrenérgicos favorece la reducción de los niveles intracelulares de AMPc, produciendo una menor activación de PKA y, por tanto, de HSL. Así, las catecolaminas tienen un efecto dual sobre la lipólisis, y su efecto neto depende del balance entre los receptores α2 y β-adrenérgicos. Otra de las hormonas inhibidoras de la lipólisis es la insulina, que induce la activación de PI3K y de la fosfodiesterasaiii, que a su vez produce la inactivación de AMPc. En adipocitos de pacientes obesos, la lipólisis basal está aumentada y falla la lipólisis estimulada por catecolaminas, además de existir un descenso en la expresión de HSL32. Se ha descrito que alteraciones en la lipólisis inducidas por catecolaminas pueden tener un papel importante en el desarrollo de la obesidad en humanos, así como sus complicaciones metabólicas y vasculares asociadas. La capacidad lipolítica parece tener un componente hereditario, aunque también puede verse afectada por el propio sobrepeso y por el grado de actividad física33. Asimismo, se han descrito defectos de la HSL en familias de obesos y polimorfismos de los genes para HSL y para los receptores adrenérgicos β2 y β3 en asociación con la obesidad humana. Los adipocitos de pacientes obesos se caracterizan por presentar niveles altos de receptores adrenérgicos α2 y un ratio elevado de receptores adrenérgicos α2/β34. Además, en modelos animales se ha demostrado una correlación positiva entre el grado de obesidad y el ratio de receptores adrenérgicos α2/β35.
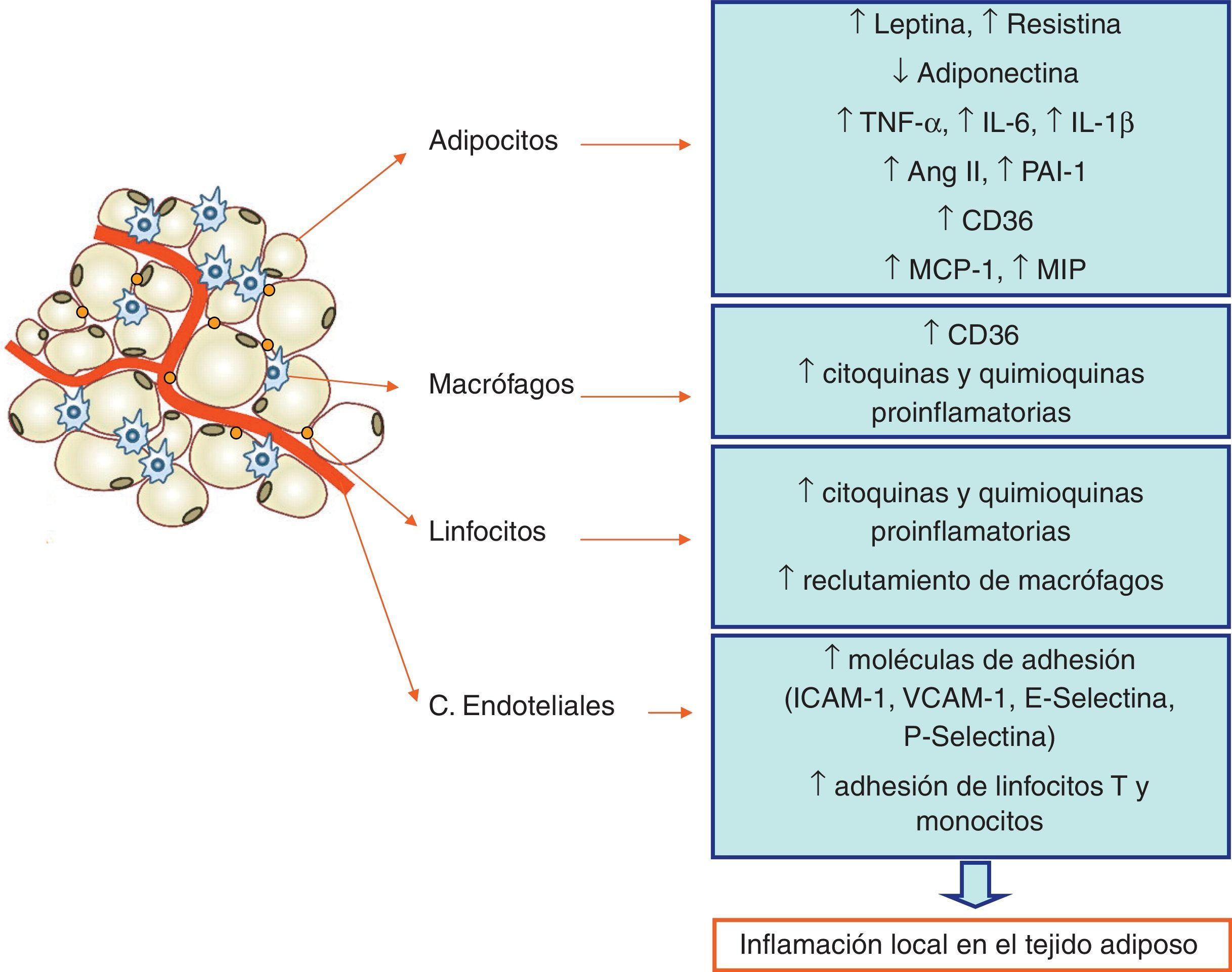
El tejido adiposo blanco como órgano endocrinoUn gran número de evidencias demuestran que el WAT, incluyendo los adipocitos, los preadipocitos y el tejido estromal, es un órgano metabólicamente activo que desempeña un papel central en la homeostasis energética. De hecho, es capaz de modificar sus propias características funcionales y morfológicas de acuerdo con las condiciones fisiológicas o patológicas predominantes. El WAT no solo es un órgano de reservorio de energía, sino también un órgano secretor de ciertas moléculas que tienen una acción endocrina, paracrina y autocrina36,37 (fig. 2).

Contribución de los distintos tipos celulares a la inflamación local del tejido adiposo. La obesidad es un estado crónico inflamatorio de bajo grado en el que tanto los adipocitos como los macrófagos y linfocitos infiltrados, así como las células endoteliales de los capilares próximos a los adipocitos, están contribuyendo a esa inflamación local.
Algunas de estas moléculas secretadas por los adipocitos están implicadas en la regulación del peso corporal (leptina, adiponectina), en la respuesta inflamatoria generada localmente en una situación de obesidad (TNF-α, IL-6 e IL-1β), en la función vascular (AngII y PAI-1) o reproductora (estrógenos, entre otras).
La leptina es una hormona secretada principalmente por los adipocitos que tiene un papel relevante en la regulación del peso corporal a través de sus efectos centrales sobre el apetito, y periféricos sobre el gasto energético38. La concentración de leptina circulante disminuye en condiciones de ayuno o restricción calórica y aumenta en respuesta a la ingesta, principalmente en respuesta a la glucosa39,40. Sin embargo, la gran mayoría de pacientes obesos presentan concentraciones elevadas de leptina y están aumentados con relación al grado de adiposidad y de hiperinsulinemia, lo que ha llevado al concepto de leptinorresistencia41. Esta hiperleptinemia ha sido involucrada en la insulinorresistencia del obeso a través de alteraciones en la fosforilación del receptor de la insulina42. Otra de las hormonas secretadas por los adipocitos que participa en el control de la ingesta es la adiponectina (Acrp30/AdipoQ). Es una adipocitoquina implicada en la regulación del metabolismo energético del organismo, ya que estimula la oxidación de ácidos grasos, reduce los triglicéridos plasmáticos y mejora el metabolismo de la glucosa mediante un aumento de la sensibilidad a la insulina43. En diversos estudios se ha observado hipoadiponectinemia en pacientes con obesidad, diabetes mellitus y arteriopatía coronaria44,45. Además de sus propiedades antidiabetogénicas, la adiponectina posee un efecto antiaterogénico y también tiene una relación inversa con otros factores de riesgo como la presión arterial, el colesterol total y las lipoproteínas de baja densidad (LDL)46-49. Los estudios transversales de población muestran que concentraciones bajas de adiponectina están relacionadas con un aumento del perfil de riesgo metabólico y cardiovascular50,51. Recientemente se ha identificado una nueva molécula, la resistina, secretada por los adipocitos maduros y que podría ser el nexo de unión entre la obesidad y el desarrollo de resistencia a la insulina52. En roedores parece estar clara su implicación en la resistencia a la insulina. Sus niveles circulantes se incrementan durante la obesidad, su bloqueo mejora la homeostasis de la glucosa y su administración ejerce un efecto negativo sobre los tejidos diana de la insulina. En humanos, sin embargo, el papel de la resistina no está ni mucho menos esclarecido, y los trabajos publicados son bastante contradictorios. Parece que esta hormona podría ejercer algún papel en la respuesta inflamatoria debido a su mayoritaria expresión en células mononucleares53. Se requieren, por tanto, nuevos estudios para determinar el papel de esta molécula tanto en la obesidad como en la resistencia a la insulina.
Diversas citoquinas proinflamatorias son secretadas por distintos tipos celulares, incluidos los adipocitos. Tienen una acción paracrina o autocrina en el propio tejido y participan en la respuesta inflamatoria local que se produce en los adipocitos de pacientes obesos. Se ha descrito que los niveles de TNF-α en el adipocito están correlacionados positivamente con el tamaño de los depósitos adiposos. Además, la expresión del ARN mensajero del TNF-α está aumentada en el tejido adiposo de distintos modelos murinos de obesidad y diabetes y de pacientes obesos, relacionándose dicho aumento con el desarrollo de resistencia a la insulina54,55. Por un lado, el TNF-α activa la lipólisis e inhibe los niveles de LPL y GLUT-4 como un mecanismo que trata de reducir el tamaño excesivo de los depósitos grasos. Sin embargo, los niveles altos de TNF-α en el tejido adiposo podrían deberse a alguna de las alteraciones metabólicas asociadas a la obesidad como es la resistencia a la insulina. Así, el TNF-α aumenta los niveles de ácidos grasos libres, reduciendo la sensibilidad a la insulina, y tiene un efecto inhibidor directo en la acción de la insulina en el hígado, incrementando la producción de glucosa hepática56,57. Sin embargo, las acciones biológicas del TNF-α podría estar influenciadas por la expresión de 2 receptores: p60TNFR (TNF-R1) y p80TNFR (TNF-R2). La expresión de TNF-R1 está positivamente correlacionada con el índice de masa corporal y el tamaño de los adipocitos, mientras que la expresión de TNF-R2 muestra asociaciones positivas con la concentración circulante de insulina y de triglicéridos58. Así, interfiriendo la vía de señalización del TNF-α se protege la resistencia a la insulina inducida por obesidad59. La neutralización del TNF-α usando un anticuerpo monoclonal reduce los niveles de glucosa del modelo murino diabético KKAy60 y mejora el control glucémico en pacientes con diabetes tipo261. Del mismo modo, el tratamiento con un anticuerpo anti-TNF-α durante 6semanas redujo la hiperglucemia en el ayuno y la intolerancia a la glucosa, y mejoró la sensibilidad a la insulina en el WAT visceral, principalmente en el compartimento gonadal, del ratón BATIRKO de 52semanas, que presenta un aumento de la adiposidad asociado a una lipoatrofia marrón severa. En este modelo, el tratamiento con anti-TNF-α redujo la activación de NF-κB en ambos tejidos adiposos, y la expresión de moléculas controladas por dicho factor de transcripción en el WAT gonadal, en el BAT y en la aorta. Además, las alteraciones vasculares, como la resistencia a la insulina vascular y la disfunción vascular, fueron revertidas por el tratamiento con anti-TNF-α62.
El angiotensinógeno y el inhibidor del activador del plasminógeno (PAI-1) son moléculas secretadas también por los adipocitos y cuya expresión génica está aumentada en situaciones de obesidad63,64, teniendo un efecto deletéreo sobre la función vascular. Además, otro componente del sistema renina-angiotensina que también tiene el adipocito es la angiotensinaii, que posee un efecto estimulante sobre la diferenciación del tejido adiposo y tiene su implicación en la regulación de la adiposidad debido a sus acciones lipogénicas65. En relación con la secreción de PAI-1 por el tejido adiposo, se ha observado una mayor producción en la grasa visceral que en la grasa subcutánea. En este sentido, los niveles de PAI-1 estaban aumentados en la obesidad central, relacionándose con las alteraciones vasculares asociados a ella66.
Otros factores importantes liberados por el WAT son los ácidos grasos libres, que se encuentran fuertemente relacionados tanto como causa y como consecuencia de la resistencia a la insulina y la diabetes tipo267,68. Los ácidos grasos interfieren en la señalización de la insulina y en el metabolismo de los hidratos de carbono66.
Por último, cabe destacar el papel de CD36 en macrófagos como un mecanismo por el cual puede vincularse la insulinorresistencia y el desarrollo de placas ateroscleróticas. Por tanto, CD36 es un péptido que pertenece a la familia de receptoresB en los macrófagos y son elementos claves en el reconocimiento, la captación y la internalización de moléculas oxidadas de LDL en los macrófagos durante las etapas iniciales del proceso aterogénico69,70. Así, en un modelo murino Ob/Ob, que es insulinorresistente, se observó un aumento de la captación de las LDL oxidadas por parte de los macrófagos, acompañado de un aumento en los niveles de CD36 y una resistencia en la vía de señalización de la insulina71. Los datos obtenidos abren una perspectiva interesante en el estudio de los mecanismos del proceso aterogénico, ya que una situación de resistencia a la insulina aumenta los niveles de CD36, y a su vez está implicada en el reconocimiento de las LDL oxidadas y en su posterior procesamiento para producir células espumosas, evento clave en la progresión de la placa aterosclerótica71.
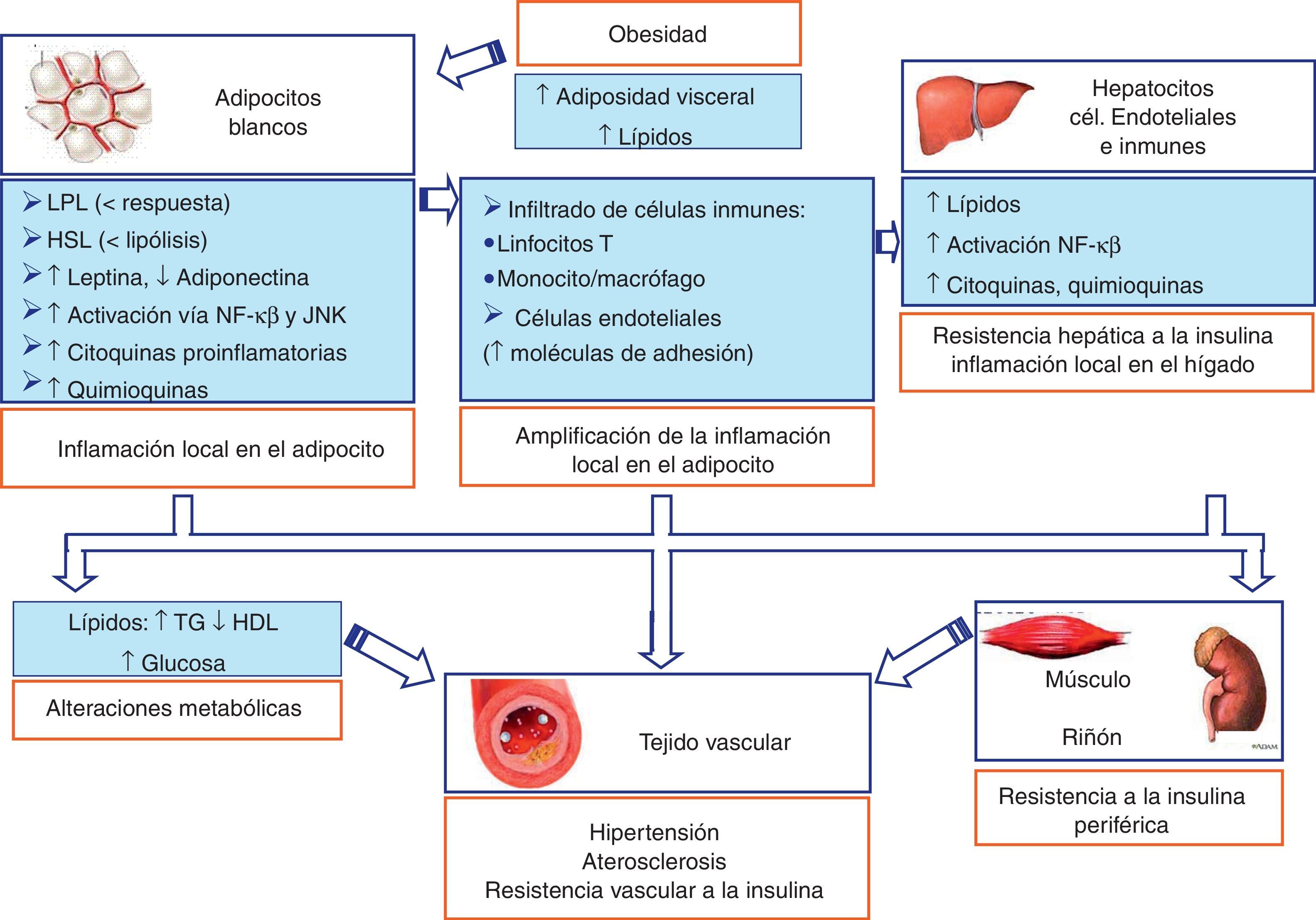
Papel del tejido adiposo blanco en la inflamación generada en la obesidad y en las alteraciones vasculares asociadasEn una situación de obesidad asociada a resistencia a la insulina, tanto el exceso de dieta como la propia obesidad producen una acumulación de lípidos en los adipocitos, iniciándose un estrés en la célula y la activación de la vía de las JNK y del factor nuclear de transcripción κB (NF-κB)72,73. Estas vías de señalización inflamatorias regulan la fosforilación de proteínas y distintos eventos celulares transcripcionales que conducen a un aumento por parte del adipocito en la producción de moléculas proinflamatorias, incluidas el TNF-α, la IL-6, la leptina y la resistina, de quimioquinas tales como la proteína quimioatractante de monocitos (MCP-1), y de otros mediadores proaterogénicos, como por ejemplo PAI-1. Moléculas de adhesión endotelial (p.ej., ICAM-1 y VCAM-1) y moléculas quimioatractantes (p.ej., CCX) se unen a integrinas y receptores de quimioquinas (CCR), respectivamente, y favorecen el reclutamiento de monocitos y otras células inflamatorias al tejido adiposo. Los monocitos, una vez en el interior, se diferencian a macrófagos y amplifican la respuesta inflamatoria produciendo muchas de las mismas citoquinas y quimioquinas inflamatorias mencionadas previamente74 (figs. 2 y 3). Un número reciente de artículos han sugerido también que los linfocitos T podrían desempeñar un papel importante en la producción de citoquinas proinflamatorias y en el reclutamiento de macrófagos al tejido adiposo en obesos. Al igual que los monocitos, las células T que circulan a través del organismo infiltran los tejidos periféricos en respuesta a las señales de quimioquinas y citoquinas. El infiltrado de linfocitos precede a la población de monocitos en respuesta a la dieta grasa y podría proporcionar mediadores proinflamatorios que promueven el reclutamiento y la activación de macrófagos (figs. 2 y 3). Los linfocitos T citotóxicos del linaje CD8 están muy enriquecidos en el tejido adiposo de ratones sometidos a dieta grasa, lo que resulta consistente con el aumento significativo de células CD8 en pacientes obesos75. Así, ratones deficientes en CD8 fueron parcialmente resistentes a desarrollar obesidad inducida por dieta rica en grasas, mientras que la transferencia de células CD8 agravaba la inflamación del tejido adiposo73 (figs. 2 y 3).
 a la obesidad y las complicaciones metabólicas y vasculares asociadas. En una situación de obesidad se produce una acumulación de lípidos en los adipocitos, iniciándose un estrés en la célula y la activación de la vía de las JNK y del NF-κB, generándose una inflamación local en el adipocito blanco. Esta puede exportarse a través de la vía portal al hígado y, finalmente, a otros tejidos periféricos, como el territorio vascular, donde podría producir aterosclerosis, hipertensión y resistencia vascular a la insulina.")
Contribución del tejido adiposo blanco (WAT) a la obesidad y las complicaciones metabólicas y vasculares asociadas. En una situación de obesidad se produce una acumulación de lípidos en los adipocitos, iniciándose un estrés en la célula y la activación de la vía de las JNK y del NF-κB, generándose una inflamación local en el adipocito blanco. Esta puede exportarse a través de la vía portal al hígado y, finalmente, a otros tejidos periféricos, como el territorio vascular, donde podría producir aterosclerosis, hipertensión y resistencia vascular a la insulina.
Así, en una situación de obesidad donde hay un exceso de grasa, en particular a nivel visceral, es capaz de crear un «ambiente inflamatorio», con incremento de TNF-α, IL-6, PAI-1, leptina, fibrinógeno y componentes del sistema renina-angiotensina-aldosterona. Esto induce y favorece una infiltración del tejido adiposo visceral por parte de los macrófagos en la obesidad76. Estos mediadores inflamatorios no solo son producidos por los adipocitos, sino también por células del sistema reticuloendotelial y por preadipocitos. La expresión de genes que codifican la síntesis de mediadores inflamatorios se encuentra aumentada en el tejido estromal adipocitario, en el que se encuentran también los macrófagos y los preadipocitos. Estos últimos también sintetizan citoquinas al ser estimulados por TNF-α77. Así, en este contexto, cambios en el tamaño del tejido adiposo inducidos por ganancia de peso generarían la secreción adipocitaria de citoquinas, lo cual induce la síntesis y la liberación de factores quimotrayentes de células del sistema reticuloendotelial por parte del tejido estromal adipocitario. El resultado final sería el infiltrado del WAT por parte de macrófagos y perpetuación de la inflamación local en el propio WAT.
Además de los propios adipocitos y las células inflamatorias, otros tipos celulares podría participar en dicha respuesta inflamatoria. Así, el tejido adiposo está vascularizado, con múltiples capilares en contacto con cada adipocito78. En este sentido, para la expansión de la grasa, la microcirculación podría desempeñar un papel clave en la inflamación del tejido adiposo. Así, los leucocitos no se adhieren en un endotelio normal que es antiadherente, mientras que después de la administración de dieta rica en grasas se ha demostrado que dicho endotelio expresa moléculas de adhesión y une leucocitos79. Las células endoteliales del tejido adiposo podrían aumentar la expresión de proteínas de adhesión, como ICAM-1, VCAM-1, E-selectina o P-selectina en respuesta a un aumento de la adiposidad y favorecer así la adhesión de células inflamatorias como linfocitos T y monocitos80.
El aumento de la adiposidad activa la respuesta inflamatoria no solo en el adipocito, sino también a través de la vía portal en el hígado81 (fig. 3). Esto sugiere que la acumulación lipídica en los hepatocitos o esteatosis podría inducir una respuesta inflamatoria subaguda en el hígado, que es similar a la inflamación en el tejido adiposo que sigue a la acumulación lipídica en el adipocito. Las moléculas proinflamatorias producidas en la grasa abdominal a través de la circulación portal podrían ser las responsables del inicio de la inflamación hepática. Además, en el propio hepatocito graso se produce la activación de NF-κB y un aumento de la expresión de citoquinas, incluidas el TNF-α, el IL-6 y el IL-1β81. Las citoquinas proinflamatorias participan en el desarrollo de la resistencia a la insulina y activan a los macrófagos hepáticos residentes, llamados células de Kupffer. El hígado está densamente poblado por células de Kupffer, que representan aproximadamente el 5% del total de las células. En la obesidad, con el aumento de la adiposidad no aumenta el número de células de Kupffer, sino su activación81. En el hígado, al igual que en el tejido adiposo, hay distintos tipos de células que participan en la inflamación y en la resistencia a la insulina a nivel local del hepatocito, como son las células inmunes y las células endoteliales82. Por tanto, los mediadores proinflamatorios y proaterogénicos que son producidos por el tejido adiposo y el hígado y están asociados a las células inmunes generan una inflamación sistémica que produce resistencia a la insulina en el músculo esquelético y otros tejidos periféricos. En el tejido vascular, además de producir resistencia a la insulina vascular, podrían contribuir a iniciar el proceso aterogénico83. La obesidad, por tanto, como un estado inflamatorio crónico de bajo grado, provee una relación directa con otros componentes del síndrome metabólico. La vía final común es la aterosclerosis, causante de enfermedad vascular generalizada que conduce a hipertensión arterial, enfermedad coronaria y enfermedad vascular periférica (fig. 3).
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.