




En este artículo se revisan las infecciones por el virus de la hepatitis B, de la hepatitis C y de la influenza, y se resumen las características más destacadas de los antivíricos empleados para el tratamiento de estas enfermedades en el adulto. Se hace referencia a la dosificación de los fármacos en función de la indicación, al ajuste de la dosis en caso de insuficiencia renal o hepática, a sus características farmacocinéticas más relevantes, así como a sus principales efectos secundarios e interacciones.
This article aims to review hepatitis B and C and influenza infections and to summarise the main characteristics of the antiviral drugs available to treat those infections in adults. The review of each drug focuses on dosage depending on treatment indication, dosage adjustment in renal or hepatic impairment, main pharmacokinetic features and the most significant adverse effects and drug interactions.
Existe un gran número de enfermedades causadas por virus que pueden afectar en mayor o menor grado a diferentes colectivos, dependiendo del estado de su sistema inmunitario. Algunas de ellas pueden ser graves, especialmente en prematuros, ancianos, trasplantados o pacientes con infección por el virus de la inmunodeficiencia humana (VIH). A continuación se revisan las infecciones causadas por el virus de la hepatitis B (VHB), de la hepatitis C (VHC) y de la influenza, así como los antivíricos utilizados en su tratamiento (tablas 1 y 2)1–6. Se describen sus características farmacocinéticas, sus efectos adversos, sus interacciones, su dosificación en el adulto en sus principales indicaciones (tabla 3)1–9 y los ajustes de dosis en situaciones de insuficiencia renal o hepática (tabla 4)1–6. Se indican asimismo los principales fármacos en investigación.
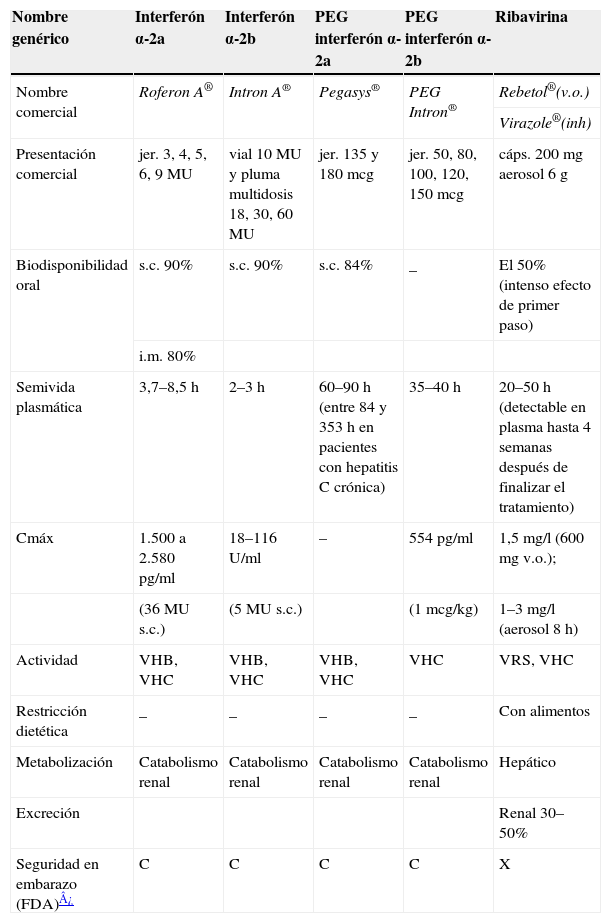
Características de los antivíricos activos frente a los virus de la hepatitis B y C
| Nombre genérico | Interferón α-2a | Interferón α-2b | PEG interferón α-2a | PEG interferón α-2b | Ribavirina |
| Nombre comercial | Roferon A® | Intron A® | Pegasys® | PEG Intron® | Rebetol®(v.o.) |
| Virazole®(inh) | |||||
| Presentación comercial | jer. 3, 4, 5, 6, 9MU | vial 10MU y pluma multidosis 18, 30, 60 MU | jer. 135 y 180mcg | jer. 50, 80, 100, 120, 150mcg | cáps. 200mg aerosol 6g |
| Biodisponibilidad oral | s.c. 90% | s.c. 90% | s.c. 84% | _ | El 50% (intenso efecto de primer paso) |
| i.m. 80% | |||||
| Semivida plasmática | 3,7–8,5h | 2–3h | 60–90h (entre 84 y 353h en pacientes con hepatitis C crónica) | 35–40h | 20–50h (detectable en plasma hasta 4 semanas después de finalizar el tratamiento) |
| Cmáx | 1.500 a 2.580pg/ml | 18–116U/ml | – | 554pg/ml | 1,5mg/l (600mg v.o.); |
| (36MU s.c.) | (5MU s.c.) | (1mcg/kg) | 1–3mg/l (aerosol 8h) | ||
| Actividad | VHB, VHC | VHB, VHC | VHB, VHC | VHC | VRS, VHC |
| Restricción dietética | _ | _ | _ | _ | Con alimentos |
| Metabolización | Catabolismo renal | Catabolismo renal | Catabolismo renal | Catabolismo renal | Hepático |
| Excreción | Renal 30–50% | ||||
| Seguridad en embarazo (FDA)¿ | C | C | C | C | X |
Cmáx: concentración plasmática máxima; i.m.: intramuscular; s.c.: subcutánea; VHB: virus de la hepatitis B; VHC: virus de la hepatitis C; v.o.: vía oral; VRS: virus respiratorio sincitial.
Categorías seguridad embarazo (FDA): A: ausencia de riesgos para el feto; B: no tiene teratogenicidad en animales, falta de estudios en humanos; C: no hay datos de seguridad en embarazadas y los estudios en animales muestran toxicidad fetal o no se han realizado, y no deben utilizarse esto fármacos a menos que el beneficio potencial supere el posible riesgo fetal; D: existe evidencia de riesgo para el feto, demostrado en embarazadas. Los beneficios terapéuticos para la madre pueden sobrepasar el riesgo según la situación; X: existe evidencia de riesgo fetal y este riesgo sobrepasa cualquier beneficio. Son medicamentos absolutamente contraindicados en el embarazo.
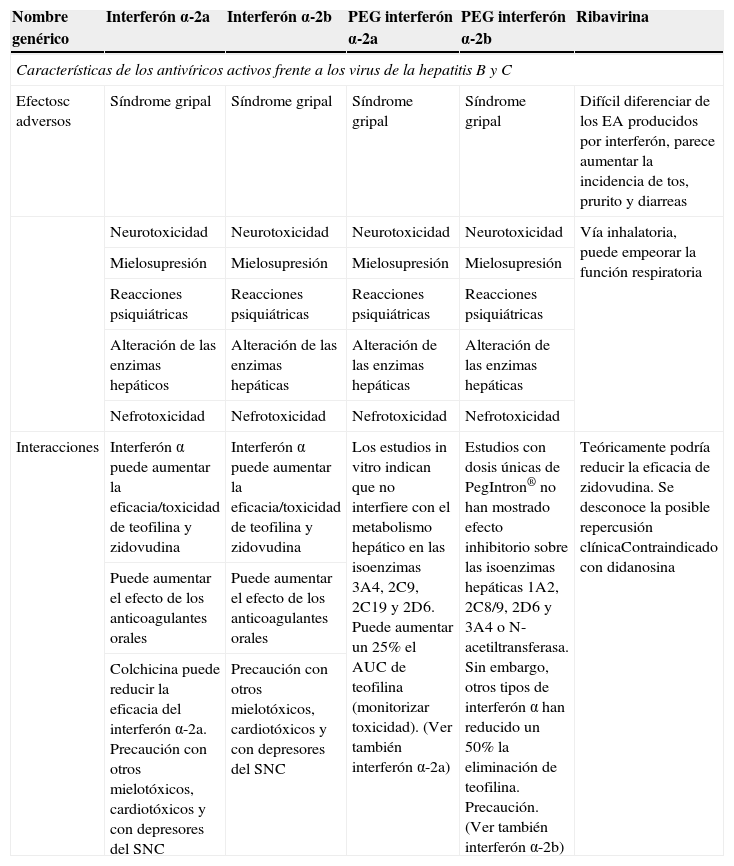
| Nombre genérico | Interferón α-2a | Interferón α-2b | PEG interferón α-2a | PEG interferón α-2b | Ribavirina |
| Características de los antivíricos activos frente a los virus de la hepatitis B y C | |||||
| Efectosc adversos | Síndrome gripal | Síndrome gripal | Síndrome gripal | Síndrome gripal | Difícil diferenciar de los EA producidos por interferón, parece aumentar la incidencia de tos, prurito y diarreas |
| Neurotoxicidad | Neurotoxicidad | Neurotoxicidad | Neurotoxicidad | Vía inhalatoria, puede empeorar la función respiratoria | |
| Mielosupresión | Mielosupresión | Mielosupresión | Mielosupresión | ||
| Reacciones psiquiátricas | Reacciones psiquiátricas | Reacciones psiquiátricas | Reacciones psiquiátricas | ||
| Alteración de las enzimas hepáticos | Alteración de las enzimas hepáticas | Alteración de las enzimas hepáticas | Alteración de las enzimas hepáticas | ||
| Nefrotoxicidad | Nefrotoxicidad | Nefrotoxicidad | Nefrotoxicidad | ||
| Interacciones | Interferón α puede aumentar la eficacia/toxicidad de teofilina y zidovudina | Interferón α puede aumentar la eficacia/toxicidad de teofilina y zidovudina | Los estudios in vitro indican que no interfiere con el metabolismo hepático en las isoenzimas 3A4, 2C9, 2C19 y 2D6. Puede aumentar un 25% el AUC de teofilina (monitorizar toxicidad). (Ver también interferón α-2a) | Estudios con dosis únicas de PegIntron® no han mostrado efecto inhibitorio sobre las isoenzimas hepáticas 1A2, 2C8/9, 2D6 y 3A4 o N-acetiltransferasa. Sin embargo, otros tipos de interferón α han reducido un 50% la eliminación de teofilina. Precaución. (Ver también interferón α-2b) | Teóricamente podría reducir la eficacia de zidovudina. Se desconoce la posible repercusión clínicaContraindicado con didanosina |
| Puede aumentar el efecto de los anticoagulantes orales | Puede aumentar el efecto de los anticoagulantes orales | ||||
| Colchicina puede reducir la eficacia del interferón α-2a. Precaución con otros mielotóxicos, cardiotóxicos y con depresores del SNC | Precaución con otros mielotóxicos, cardiotóxicos y con depresores del SNC | ||||
| Nombre genérico | Adefovir dipivoxilo | Emtricitabina | Entecavir | Lamivudina 3TC | Telbivudina | Tenofovir |
| Nombre comercial | Hepsera® | Emtriva® | Baraclude® | Zeffix® | Sebivo® | Viread® |
| Presentación comercial | comp. 10mg | comp. 200mg y solución 10mg/ml | comp. 0,5 y 1 mg | comp. 100mg y solución 5mg/ml | comp. 600mg | comp. 245mg |
| Biodisponibilidad oral | 59% | 93% (cápsulas) 75% (solución) | Se estima que es al menos del 70% | 85% | -- | El 25% en ayunas (con alimentos grasos AUC 40% mayor) |
| Semivida plasmática | 7,22h (4,72–10,70h) | 10h | 128–149h | 5–7h (lamivudina trifosfato en los hepatocitos in vitro: 17–19h) | 41,8±11,8h | En plasma: 12–18h |
| Tenofovir difosfato intracelular en PBMC activadas 10h; en PBMC en reposo 50h | ||||||
| Cmáx (±DE) | 17,8±3,2ng/ml | 1,8±0,7mcg/ml | 4,2ng/ml (0,5mg) | 1,1–1,5mcg/ml | 3,2±1,1mcg/ml | 326ng/ml |
| 8,2ng/ml (1mg) | ||||||
| Actividad | VHB | VIH, VHB (solamente se encuentra autorizada la indicación de VIH) | VHB | VHB/VIH-1,2 | VHB | VIH, VHB (solamente se encuentra autorizada la indicación de VIH) |
| Restricción dietética | No | No | No | No | No | Con alimentos |
| Metabolización | No | Escasa (no a través del citocromo P450) | Escasa (no a través del citocromo P450) | Escasa (5–10%) | No | No |
| Excreción | Vía renal (filtración glomerular y secreción tubular activa). El 45% de la dosis se recupera en forma de adefovir en la orina de 24h | Renal (86% principalmente inalterado, 13% metabolitos). El 14% por heces | Vía renal aprox. el 75% inalterado, filtración glomerular y secreción tubular activa | Vía renal aprox. el 70% inalterado, filtración glomerular y secreción tubular activa (sistema de transporte catiónico orgánico) | Vía renal como fármaco inalterado, por filtración glomerular | Vía renal aprox. el 70–80% inalterado, filtración glomerular y secreción tubular activa (en la que intervienen los transportadores hOAT 1 y 3 y MRP4) |
| Seguridad en embarazo (FDA)¿ | C | B | C | C | B | B |
| Efectos adversos | Insuficiencia renal, hipofosfatemia | Cefalea, molestias digestivas, aumento de CPK | Cefalea, astenia, molestias digestivas, elevación de aminotransferasas | Cefalea, astenia, molestias digestivas | Cefalea, astenia, molestias digestivas, tos, elevación de aminotransferasas | Hipofosfatemia, mareo, molestias digestivas |
| En <1%, insuficiencia renal | ||||||
| Interacciones | No es de esperar que tenga interacciones a nivel de metabolismo | No es de esperar que tenga interacciones a nivel de metabolismo | No es de esperar que tenga interacciones a nivel de metabolismo | No es de esperar que tenga interacciones a nivel de metabolismo | No es de esperar que tenga interacciones a nivel de metabolismo | No es de esperar que tenga interacciones a nivel de metabolismo |
| Ausencia de interacción con lamivudina Puede interaccionar con otros medicamentos que se eliminen mediante secreción tubular o por el transportador de aniones orgánicos hOAT1 (cidofovir) Precaución con nefrotóxicos. Evitar asociar a tenofovir | Puede interaccionar con otros medicamentos que se eliminen mediante secreción tubular activa o empeoren la función renal. No se recomienda su asociación con lamivudina por falta de experiencia clínica | Puede interaccionar con otros medicamentos que se eliminen mediante secreción tubular activa o empeoren la función renal | Puede interaccionar con otros medicamentos que se eliminen mediante secreción tubular activa o empeoren la función renal. Cotrimoxazol aumenta un 40% el AUC de lamivudina (sólo requiere ajuste de dosis en caso de insuf. renal) | Puede interaccionar con medicamentos que alteren la función renal. Ausencia de interacción con lamivudina, adefovir, tenofovir o ciclosporina. En un ensayo clínico, se observó un mayor riesgo de desarrollar neuropatía con interferón α-2a pegilado (180 mcg/semana) junto con telbivudina | Puede interaccionar con medicamentos que alteren la función renal o empleen alguno de los transportadores, como p. ej. cidofovir. Ausencia de interacción farmacocinética con adefovir, emtricitabina, lamivudina o ribavirina | |
| No se han observado interacciones farmacocinéticas entre entecavir y lamivudina, adefovir o tenofovir | Ausencia de interacción con interferón α, ciclosporina y entecavir. No se recomienda su asociación con emtricitabina por falta de experiencia clínica | Evitar asociar a adefovir y a otros nefrotóxicos. (En pacientes que reciben tratamiento antirretrovírico cabe considerar otras interacciones adicionales: consultar ficha técnica del producto) |
| Nombre genérico | Amantadina | Zanamivir | Oseltamivir |
| Características de los antivíricos activos frente a virus de la influenza | |||
| Nombre comercial | Amantadina Llorente® | Relenza® | Tamiflu® |
| Amantadina Belmac® | |||
| Presentación comercial | caps. 100mg | Rotadisk | caps. 75mg |
| 4alv/5mg | susp. oral 12mg/ml (comercial) | ||
| susp. oral 15mg/ml (fórmula magistral a partir de sustancia) | |||
| Biodisponibilidad oral | 85–90% | Oral 2%. Inhalada 4–25% | 75% |
| Vida media plasmática | 12–18h | 2,5–5,1h | Fosfato (profármaco): 1–3h |
| Carboxilato (forma activa): 6–10h | |||
| Cmáx | 0,5–0,8mg/l (200mg v.o.) | _ | Fosfato (profármaco): 65,2ng/ml |
| Carboxilato (forma activa): 348ng/ml | |||
| CI 50/90% | 90: <1mg/l | _ | |
| Actividad | Influenza-A | Influenza A y B | Influenza A y B |
| Restricción dietética | No | ‐ | No |
| Metabolización | No | No | El 90% metabolismo hepático de fosfato de oseltamivir a carboxilato de oseltamivir |
| Excreción | Eliminación renal mayoritaria (90% fármaco activo) | Eliminación renal mayoritaria (>90% del fármaco activo absorbido). El no absorbido se deposita en la orofaringe y se elimina a través de las heces | Eliminación renal mayoritaria (90% fármaco activo) |
| Seguridad en embarazo (FDA)¿ y lactancia | Embriotóxico y teratógeno en animales. Contraindicado. Se elimina a través de la leche materna | C | C |
| Efectos adversos | EA a nivel del SNC (nerviosismo, ansiedad, insomnio, confusión) | Bien tolerado | Bien tolerado |
| Alteraciones GI | Incidencia de efectos adversos similar a placebo | Incidencia de efectos adversos similar a placebo | |
| Edemas periféricos | Raramente broncoespasmo (pacientes con asma o EPOC) | ||
| Interacciones | Puede potenciar los EA a nivel del SNC de otros fármacos anticolinérgicos | _ | _ |
| Triamtereno aumenta la toxicidad de amantadina | |||
AUC: área bajo la curva de niveles plasmáticos; CI: concentración inhibitoria; Cmáx: concentración plasmática máxima; CPK: creatina-fosfocinasa; DE: desviación estándar; EA: efectos adversos; EPOC: enfermedad pulmonar obstructiva crónica; GI: gastrointestinal; PBMC: células mononucleares de sangre periférica; SNC: sistema nervioso central; VHB: virus de la hepatitis B; VIH: virus de la inmunodeficiencia humana; v.o.: vía oral.
Categorías seguridad embarazo (FDA): A: ausencia de riesgos para el feto; B: no tiene teratogenicidad en animales, falta de estudios en humanos; C: no hay datos de seguridad en embarazadas y los estudios en animales muestran toxicidad fetal o no se han realizado, y no deben utilizarse esto fármacos a menos que el beneficio potencial supere el posible riesgo fetal. D: existe evidencia de riesgo para el feto, demostrado en embarazadas. Los beneficios terapéuticos para la madre pueden sobrepasar el riesgo según la situación; X: existe evidencia de riesgo fetal y este riesgo sobrepasa cualquier beneficio. Son medicamentos absolutamente contraindicados en el embarazo.
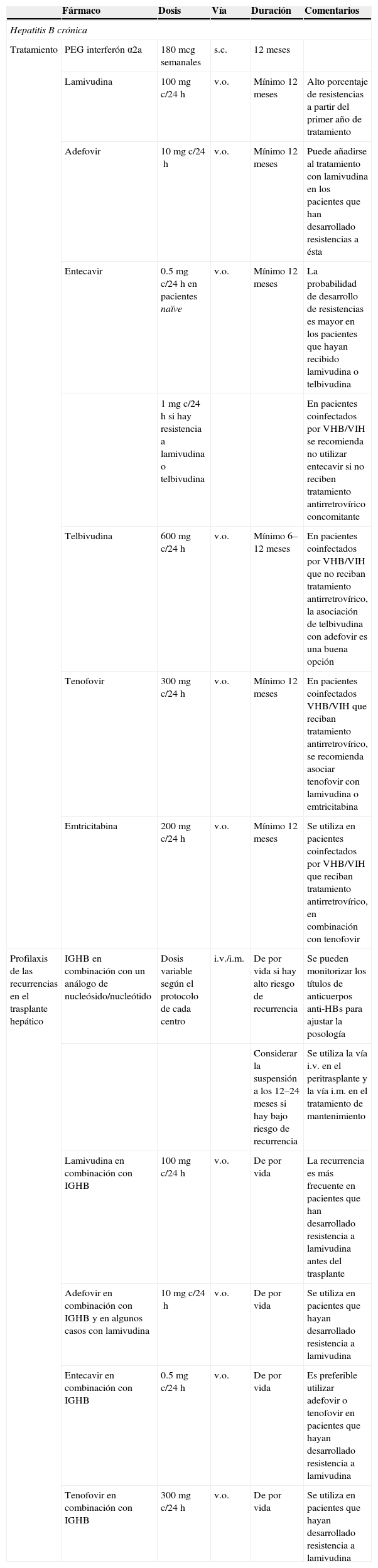
Infección por el virus de la hepatitis B y C y el virus de la influenza: dosificación de los antivíricos en pacientes adultos con función renal normal (si hay insuficiencia renal, ver tabla 4)
| Fármaco | Dosis | Vía | Duración | Comentarios | |
| Hepatitis B crónica | |||||
| Tratamiento | PEG interferón α2a | 180mcg semanales | s.c. | 12 meses | |
| Lamivudina | 100mg c/24h | v.o. | Mínimo 12 meses | Alto porcentaje de resistencias a partir del primer año de tratamiento | |
| Adefovir | 10mg c/24h | v.o. | Mínimo 12 meses | Puede añadirse al tratamiento con lamivudina en los pacientes que han desarrollado resistencias a ésta | |
| Entecavir | 0.5mg c/24h en pacientes naïve | v.o. | Mínimo 12 meses | La probabilidad de desarrollo de resistencias es mayor en los pacientes que hayan recibido lamivudina o telbivudina | |
| 1mg c/24h si hay resistencia a lamivudina o telbivudina | En pacientes coinfectados por VHB/VIH se recomienda no utilizar entecavir si no reciben tratamiento antirretrovírico concomitante | ||||
| Telbivudina | 600mg c/24h | v.o. | Mínimo 6–12 meses | En pacientes coinfectados por VHB/VIH que no reciban tratamiento antirretrovírico, la asociación de telbivudina con adefovir es una buena opción | |
| Tenofovir | 300mg c/24h | v.o. | Mínimo 12 meses | En pacientes coinfectados VHB/VIH que reciban tratamiento antirretrovírico, se recomienda asociar tenofovir con lamivudina o emtricitabina | |
| Emtricitabina | 200mg c/24h | v.o. | Mínimo 12 meses | Se utiliza en pacientes coinfectados por VHB/VIH que reciban tratamiento antirretrovírico, en combinación con tenofovir | |
| Profilaxis de las recurrencias en el trasplante hepático | IGHB en combinación con un análogo de nucleósido/nucleótido | Dosis variable según el protocolo de cada centro | i.v./i.m. | De por vida si hay alto riesgo de recurrencia | Se pueden monitorizar los títulos de anticuerpos anti-HBs para ajustar la posología |
| Considerar la suspensión a los 12–24 meses si hay bajo riesgo de recurrencia | Se utiliza la vía i.v. en el peritrasplante y la vía i.m. en el tratamiento de mantenimiento | ||||
| Lamivudina en combinación con IGHB | 100mg c/24h | v.o. | De por vida | La recurrencia es más frecuente en pacientes que han desarrollado resistencia a lamivudina antes del trasplante | |
| Adefovir en combinación con IGHB y en algunos casos con lamivudina | 10mg c/24h | v.o. | De por vida | Se utiliza en pacientes que hayan desarrollado resistencia a lamivudina | |
| Entecavir en combinación con IGHB | 0.5mg c/24h | v.o. | De por vida | Es preferible utilizar adefovir o tenofovir en pacientes que hayan desarrollado resistencia a lamivudina | |
| Tenofovir en combinación con IGHB | 300mg c/24h | v.o. | De por vida | Se utiliza en pacientes que hayan desarrollado resistencia a lamivudina | |
| Fármaco | Dosis | Vía | Duración | Comentarios | |
| Hepatitis C crónica | |||||
| Pacientes en los que la ribavirina está contraindicada o no la toleran | PEG interferón α-2b en monoterapia3 | 0,5–1mg/kg semanales | s.c. | 12 meses | |
| PEG interferón α-2a monoterapia3 | 180mcg semanales | s.c. o i.m. | 12 meses | ||
| Tratamiento combinado de ribavirina con interferón o peginterferón α | Interferón α-2b monoterapia3 | 3MU 3 veces por semana | s.c. o i.m. | 12–18 meses (ver texto) | |
| Interferón α-2a monoterapia3 | 3–6MU 3 veces por semana durante 6 meses, seguido de 3MU 3 veces por semana 6 meses más. | s.c. o i.m. | 12 meses (ver texto) | ||
| PEG interferón α-2b terapia combinada | 1,5mcg/kg semanales | s.c. | 12–24 meses (ver texto) | ||
| PEG interferón α-2a terapia combinada | 180mcg semanales | s.c. | 6–12 meses (ver texto) | ||
| Tratamiento combinado de ribavirina con interferón o peginterferón α | Ribavirina (Rebetol®) | <65kg: 400mg desayuno y 400mg cena | v.o. | 6–12 meses (ver texto) | |
| en combinación con interferón α-2b | 65–85kg: 400mg desayuno y 600mg cena | ||||
| 85–105kg: 600 desayuno y cena | |||||
| >105kg: 600mg desayuno y 800mg cena | |||||
| Ribavirina (Copegus®) en | ≤75kg: 400mg desayuno y 600mg cena | v.o. | 6–12 meses (ver texto) | ||
| combinación con interferón α-2a | >75kg: 600mg desayuno y cena | ||||
| Influenza | |||||
| Tratamiento | Amantadina | 100mg c/12h | v.o. | Hasta 24–48h de desaparición de los síntomas | No es activa sobre el virus de la influenza A (H3N2) |
| Zanamivir | 2 inh (=10mg) c/12h | v.o. | 5 días | Activo sobre el virus de la influenza A y B | |
| Oseltamivir | 75mg c/12h | v.o. | 5 días | Activo sobre el virus de la influenza A y B, pero algunas cepas del virus A (H1N1) son resistentes. El nuevo virus de la gripe A (H1N1) detectado inicialmente en México en abril del 2009 es sensible a este antivírico | |
| Profilaxis | Amantadina | 100mg c/12h | v.o. | Hasta 2 semanas después de la vacunación, o durante el período de riesgo de gripe en pacientes no vacunados | |
| Zanamivir | 2 inh (=10mg) c/24h | v.o. | Postexposición: 10 días; | ||
| durante un brote en la población: hasta 28 días | |||||
| Oseltamivir | 75mg c/24h | v.o. | Postexposición: 10 días; | ||
| durante un brote en la población: hasta 42 días | |||||
IGHB: inmunoglobulina específica antihepatitis B; i.m.: intramuscular; i.v.: intravenoso; PEG: pegilado; s.c.: subcutáneo; VHB: virus de la hepatitis B; VIH: virus de la inmunodeficiencia humana; v.o.: vía oral.
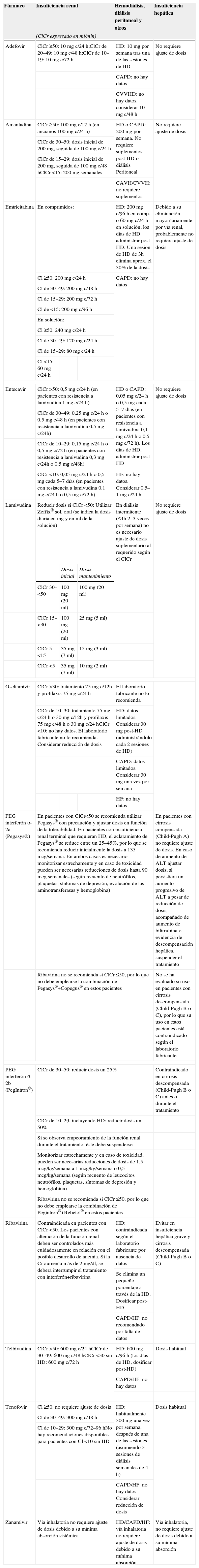
Ajustes de dosis en pacientes adultos con insuficiencia renal
| Fármaco | Insuficiencia renal | Hemodiálisis, diálisis peritoneal y otros | Insuficiencia hepática | ||
| (ClCr expresado en ml/min) | |||||
| Adefovir | ClCr ≥50: 10mg c/24h;ClCr de 20–49: 10mg c/48h;ClCr de 10–19: 10mg c/72h | HD: 10mg por semana tras una de las sesiones de HD | No requiere ajuste de dosis | ||
| CAPD: no hay datos | |||||
| CVVHD: no hay datos, considerar 10mg c/48h | |||||
| Amantadina | ClCr ≥50: 100mg c/12h (en ancianos 100mg c/24h) | HD o CAPD: 200mg por semana. No requiere suplementos post-HD o diálisis Peritoneal | No requiere ajuste de dosis | ||
| ClCr de 30–50: dosis inicial de 200mg, seguida de 100mg c/24h | |||||
| ClCr de 15–29: dosis inicial de 200mg, seguida de 100mg c/48hClCr <15: 200mg semanales | |||||
| CAVH/CVVH: no requiere suplementos | |||||
| Emtricitabina | En comprimidos: | HD: 200mg c/96h en comp. o 60mg c/24h en solución; los días de HD administrar post-HD. Una sesión de HD de 3h elimina aprox. el 30% de la dosis | Debido a su eliminación mayoritariamente por vía renal, probablemente no requiera ajuste de dosis | ||
| Cl ≥50: 200mg c/24h | CAPD: no hay datos | ||||
| Cl de 30–49: 200mg c/48h | |||||
| Cl de 15–29: 200mg c/72h | |||||
| Cl de <15: 200mg c/96h | |||||
| En solución: | |||||
| Cl ≥50: 240mg c/24h | |||||
| Cl de 30–49: 120mg c/24h | |||||
| Cl de 15–29: 80mg c/24h | |||||
| Cl <15: 60mg c/24h | |||||
| Entecavir | ClCr >50: 0,5mg c/24h (en pacientes con resistencia a lamivudina 1mg c/24h) | HD o CAPD: 0,05mg c/24h o 0,5mg cada 5–7 días (en pacientes con resistencia a lamivudina 0,1mg c/24h o 0,5mg c/72h). Los días de HD, administrar post-HD | No requiere ajuste de dosis | ||
| ClCr de 30–49: 0,25mg c/24h o 0,5mg c/48h (en pacientes con resistencia a lamivudina 0,5mg c/24h) | |||||
| ClCr de 10–29: 0,15mg c/24h o 0,5mg c/72h (en pacientes con resistencia a lamivudina 0,3mg c/24h o 0,5mg c/48h) | |||||
| ClCr <10: 0,05mg c/24h o 0,5mg cada 5–7 días (en pacientes con resistencia a lamivudina 0,1mg c/24h o 0,5mg c/72h) | HF: no hay datos. Considerar 0,5–1mg c/24h | ||||
| Lamivudina | Reducir dosis si ClCr <50: Utilizar Zeffix® sol. oral (se indica la dosis diaria en mg y en ml de la solución) | En diálisis intermitente (≤4h 2–3 veces por semana) no es necesario ajuste de dosis suplementario al requerido según el ClCr | No requiere ajuste de dosis | ||
| Dosis inicial | Dosis mantenimiento | ||||
| ClCr 30–<50 | 100mg (20ml) | 100mg (20ml) | |||
| ClCr 15–<30 | 100mg (20ml) | 25mg (5ml) | |||
| ClCr 5–<15 | 35mg (7ml) | 15mg (3ml) | |||
| ClCr <5 | 35mg (7ml) | 10mg (2ml) | |||
| Oseltamivir | ClCr >30: tratamiento 75mg c/12h y profilaxis 75mg c/24h | El laboratorio fabricante no lo recomienda | |||
| ClCr de 10–30: tratamiento 75mg c/24h o 30mg c/12h y profilaxis 75mg c/48h o 30mg c/24hClCr <10: no hay datos. El laboratorio fabricante no lo recomienda. Considerar reducción de dosis | HD: datos limitados. Considerar 30mg post-HD (administrándolo cada 2 sesiones de HD) | ||||
| CAPD: datos limitados. Considerar 30mg una vez por semana | |||||
| HF: no hay datos | |||||
| PEG interferón α-2a (Pegasys®) | En pacientes con ClCr<50 se recomienda utilizar Pegasys® con precaución y ajustar dosis en función de la tolerabilidad. En pacientes con insuficiencia renal terminal que requieran HD, el aclaramiento de Pegasys® se reduce entre un 25–45%, por lo que se recomienda reducir inicialmente la dosis a 135 mcg/semana. En ambos casos es necesario monitorizar estrechamente y en caso de toxicidad pueden ser necesarias reducciones de dosis hasta 90mcg semanales (según recuento de neutrófilos, plaquetas, síntomas de depresión, evolución de las aminotransferasas y hemoglobina) | En pacientes con cirrosis compensada (Child-Pugh A) no requiere ajuste de dosis. En caso de aumento de ALT ajustar dosis; si persistiera un aumento progresivo de ALT a pesar de reducción de dosis, acompañado de aumento de bilirrubina o evidencia de descompensación hepática, suspender el tratamiento | |||
| Ribavirina no se recomienda si ClCr ≤50, por lo que no debe emplearse la combinación de Pegasys®+Copegus® en estos pacientes | No se ha evaluado su uso en pacientes con cirrosis descompensada (Child-Pugh B o C), por lo que su uso en estos pacientes está contraindicado según el laboratorio fabricante | ||||
| PEG interferón α-2b (PegIntron®) | ClCr de 30–50: reducir dosis un 25% | Contraindicado en cirrosis descompensada (Child-Pugh B o C) antes o durante el tratamiento | |||
| ClCr de 10–29, incluyendo HD: reducir dosis un 50% | |||||
| Si se observa empeoramiento de la función renal durante el tratamiento, éste debe suspenderse | |||||
| Monitorizar estrechamente y en caso de toxicidad, pueden ser necesarias reducciones de dosis de 1,5mcg/kg/semana a 1mcg/kg/semana o 0,5mcg/kg/semana (según recuento de leucocitos neutrófilos, plaquetas, síntomas de depresión y hemoglobina) | |||||
| Ribavirina no se recomienda si ClCr ≤50, por lo que no debe emplearse la combinación de Pegintron®+Rebetol® en estos pacientes | |||||
| Ribavirina | Contraindicada en pacientes con ClCr <50. Los pacientes con alteración de la función renal deben ser controlados más cuidadosamente en relación con el posible desarrollo de anemia. Si la Cr aumenta más de 2mg/dl, se deberá interrumpir el tratamiento con interferón+ribavirina | HD: contraindicada según el laboratorio fabricante por ausencia de datos | Evitar en insuficiencia hepática grave y cirrosis descompensada (Child-Pugh B o C) | ||
| Se elimina un pequeño porcentaje a través de la HD. Dosificar post-HD | |||||
| CAPD/HF: no recomendado por falta de datos | |||||
| Telbivudina | ClCr >50: 600mg c/24hClCr de 30–49: 600mg c/48hClCr <30 sin HD: 600mg c/72h | HD: 600mg c/96h (los días de HD, dosificar post-HD) | Dosis habitual | ||
| CAPD/HF: no hay datos | |||||
| Tenofovir | Cl ≥50: no requiere ajuste de dosis | HD: habitualmente 300mg una vez por semana, después de una de las sesiones (asumiendo 3 sesiones de diálisis semanales de 4h) | Dosis habitual | ||
| Cl de 30–49: 300mg c/48h | |||||
| Cl de 10–29: 300mg c/72–96hNo hay recomendaciones disponibles para pacientes con Cl <10 sin HD | |||||
| CAPD/HF: no hay datos. Considerar reducción de dosis | |||||
| Zanamivir | Vía inhalatoria no requiere ajuste de dosis debido a su mínima absorción sistémica | HD/CAPD/HF: vía inhalatoria no requiere ajuste de dosis debido a su mínima absorción | Vía inhalatoria, no requiere ajuste de dosis debido a su mínima absorción | ||
ALT: alaninoaminotransferasa; CAPD: diálisis peritoneal continua ambulatoria; CAVH: hemofiltración continua arteriovenosa; Cl: aclaramiento; ClCr: aclaramiento de creatinina; Cr: creatinina sérica; CVVH: hemofiltración continua venovenosa; CVVHD: hemodiálisis continua venovenosa; HD: hemodiálisis (estándar); HF: hemofiltración; PEG: pegilado.
*Aclaramiento de creatinina expresado en ml/min, salvo que se indique lo contrario.
En este artículo se actualiza la revisión publicada en esta revista en el año 200310, por lo que se indican tan solo las citas bibliográficas nuevas. Para la actualización de esta revisión se ha realizado una búsqueda en PubMed (2003–2009) utilizando los términos «Hepatitis B», «Hepatitis C», «Influenza, Human» y «drug therapy [subheading]», limitando la búsqueda a «Meta-Analysis», «Randomized Controlled Trial» o «Review» publicados en lengua inglesa o castellana. Adicionalmente, se han seleccionado las revisiones de la Cochrane library relacionadas con el tema, así como la existencia de guías de la Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica o de la Infectious Diseases Society of America.
Antivíricos activos frente al virus de la hepatitis BEn la actualidad, aproximadamente 400 millones de individuos en el mundo son portadores crónicos del VHB11. De ellos, es de esperar que entre un 15–40% desarrollen complicaciones hepáticas (fallo hepático, cirrosis y hepatocarcinoma), que son consecuencia de la actividad del sistema inmunitario frente a la infección. La prevalencia de la enfermedad es muy variable entre las distintas áreas geográficas y va desde el 0,1–20%. En Europa, se estima que el 0,2–1% de la población tiene hepatitis B crónica. Las formas más frecuentes de contagio en nuestro país son principalmente el contacto sexual y el uso de drogas por vía parenteral12, a diferencia de los países con más prevalencia, donde predomina la transmisión vertical o bien la horizontal durante la infancia. La hepatitis B aguda es una enfermedad que cura espontáneamente en la mayoría de los adultos infectados, y evoluciona a hepatitis crónica en menos del 2% de los casos. Contrariamente, cuando la infección se adquiere durante el primer año de vida (ya sea por transmisión vertical u horizontal), esta se convierte en crónica en la mayoría de pacientes.
La hepatitis vírica es una enfermedad heterogénea en la que tanto la necesidad de administrar tratamiento como la eficacia de este pueden variar de unos pacientes a otros, por lo que las decisiones sobre la conducta terapéutica son a menudo difíciles y debe tomarlas un especialista. En muchos casos, sigue un curso favorable, con remisión espontánea y duradera, en algunos casos se mantiene estable durante mucho tiempo y en otros casos el curso puede ser desfavorable, hacia la transformación cirrótica del hígado que, eventualmente, puede complicarse con la aparición de un carcinoma hepatocelular. Con los tratamientos actuales es muy difícil conseguir la erradicación total del virus. Sólo el 3–8% de los pacientes que se tratan con interferón (IFN) consiguen una respuesta considerada completa, reflejada por la eliminación del antígeno de superficie del VHB y la aparición de anticuerpos anti-HBs. Este porcentaje es aun menor (<2%) en los pacientes tratados con análogos de nucleósidos13. Además, se considera que, aunque se haya producido la seroconversión, el VHB puede reaparecer aprovechando períodos de inmunodepresión. Esto se debe a que persiste en reservorios extrahepáticos, puede integrarse en el genoma humano y se encuentra en forma de ADN circular (ccADN) en los hepatocitos. Por tanto, el principal objetivo del tratamiento se dirige a la supresión de la replicación vírica de forma mantenida para evitar la aparición a largo plazo de complicaciones hepáticas (cirrosis y hepatocarcinoma) y aumentar así la supervivencia.
La decisión de cuándo iniciar el tratamiento de la hepatitis B crónica se basa en los siguientes parámetros: presencia del antígeno e de la hepatitis B (HBeAg), número de copias del ADN del VHB por PCR, niveles de alaninoaminotransferasa (ALT) y resultados de la biopsia hepática. Actualmente disponemos de 3 grupos de fármacos para el tratamiento de la hepatitis B crónica: el IFN-α-2a, el IFN-α-2b y el IFN pegilado (PEG-IFN) α-2a, los análogos de nucleósidos (lamivudina, emtricitabina, entecavir y telbivudina) y los análogos de nucleótidos (tenofovir y adefovir). Cabe destacar que la emtricitabina y el PEG-IFN-α-2b no disponen de la indicación en nuestro país. Por otro lado, carecemos de estudios bien diseñados sobre la eficacia del tratamiento combinado de antivíricos en la hepatitis B. Los pocos estudios realizados hasta el momento no han demostrado diferencias significativas frente a la monoterapia en pacientes sin tratamiento previo. Por este motivo, las guías de tratamiento aconsejan utilizar únicamente combinaciones de fármacos con la finalidad de evitar la aparición de complicaciones hepáticas graves en pacientes que han desarrollado resistencia a alguno de los medicamentos13. El uso de IFN se ve limitado por la toxicidad y el de lamivudina, telbivudina y adefovir por la aparición de resistencias. El tenofovir y el entecavir poseen una alta barrera genética y se consideran las mejores opciones de tratamiento de primera línea por vía oral. A continuación se describen los principales fármacos utilizados en el tratamiento de la hepatitis B, así como los nuevos fármacos en investigación.
Interferón αEl IFN es una sustancia natural que sintetizan las células infectadas por virus para proteger de la infección a las células sanas de la misma especie. Además de actividad antivírica, los IFN poseen acción antiproliferativa e inmunomoduladora, por lo que son también de utilidad en el tratamiento de algunos tumores. Diferentes estudios han mostrado la eficacia de los IFN en enfermedades víricas como la hepatitis B y C, por el VHS, el virus de la varicelazóster, el CMV, el rinovirus y el papilomavirus. El IFN-α puede producir también algún beneficio en pacientes con infección por el virus de la hepatitis tipo D o G.
En aproximadamente una tercera parte de los adultos y los niños con hepatitis B crónica, el IFN-α da lugar a la desaparición del HBeAg, la normalización de las aminotransferasas, la mejoría histológica mantenida y, en los adultos, al menor riesgo de hepatopatía progresiva. Los factores que determinan una mayor probabilidad de pérdida de HBeAg son niveles altos de ALT, baja viremia del VHB y presencia de actividad inflamatoria en la biopsia hepática.
Los pacientes coinfectados por el VIH y el VHB suelen responder peor al tratamiento con IFN14 que los pacientes infectados únicamente por el VHB. Sin embargo, el PEG-IFN es una de las opciones de primera elección en los pacientes coinfectados en los que no está indicado el inicio de tratamiento antirretrovírico o en los pacientes bien controlados con una combinación de antiretrovíricos que no es activa frente al VHB. Se aconseja que los pacientes candidatos a tratamiento con IFN tengan un recuento de linfocitos CD4 superior a 500 células/μl13.
La hepatitis D solo se desarrolla en pacientes infectados por el VHB y puede responder únicamente al tratamiento con dosis altas de IFN convencional o bien PEG-IFN-α-2a, pero con frecuentes recidivas. Por esto el tratamiento debe durar al menos 48 semanas y algunos autores aconsejan prolongarlo en los pacientes respondedores, adecuando la dosis a la tolerancia del paciente15. Los análogos de nucleósidos no han demostrado actividad frente al virus de la hepatitis D.
Actualmente, el PEG-IFN-α-2a (Pegasys®) ha sustituido al IFN-α-2a y al IFN-α-2b en el tratamiento de la hepatitis B. Los ensayos clínicos demuestran que el PEG-IFN-α-2a tiene una eficacia comparable o ligeramente superior a los IFN no pegilados y, además, posee la ventaja de una mayor comodidad de administración16. La dosis establecida de PEG-IFN-α-2a para el tratamiento de la hepatitis B es de 180mcg una vez a la semana, aunque dosis inferiores (entre 90–180mcg) podrían ser igualmente efectivas según datos de los estudios de fase ii16. En caso de toxicidad, las dosis deben reducirse o se debe suspender el tratamiento (ver apartado sobre hepatitis C).
La duración recomendada del tratamiento con IFN es de 48 semanas, sin embargo, los ensayos clínicos en fases ii y iii indican que 24 semanas podrían ser suficientes16,17. Si se confirmara esta opción mediante estudios convenientemente diseñados, la reducción de la duración del tratamiento podría beneficiar a pacientes con mayor probabilidad de respuesta, como los que presentan HBeAg positivo o genotipos A y B. Por otro lado, también sería necesario determinar si la prolongación del tratamiento por encima de las 48 semanas mejoraría la duración de la respuesta mantenida en pacientes con características menos favorables (HBeAg negativo o genotipo D), tal y como demuestran algunos estudios con pocos pacientes18,19. Hasta el momento se prefiere tratar a los pacientes con HBeAg negativo con fármacos alternativos al IFN por la tendencia de la enfermedad a recidivar una vez suspendido el tratamiento. Estos pacientes requieren tratamientos largos que, por tanto, presenten un buen perfil de seguridad con pocos efectos adversos.
El IFN-α actúa a través de 2 mecanismos: la inhibición directa de la replicación intracelular del virus y la estimulación de la actividad lítica de los linfocitos citotóxicos frente a las células hepáticas infectadas. Ello puede inducir un aumento transitorio de la necrosis hepatocelular que generalmente beneficia a los pacientes con hepatitis crónica simple, pero puede precipitar descompensación de la enfermedad en pacientes con cirrosis ya establecida. Por este motivo, el IFN está contraindicado en pacientes cirróticos y en la enfermedad hepática descompensada.
LamivudinaLa lamivudina fue el primer análogo de nucleósidos indicado para el tratamiento de la hepatitis B crónica y ha sido considerado el fármaco de referencia durante años. En los pacientes con HBeAg positivo y niveles elevados de ALT, el tratamiento con lamivudina consiguió la seroconversión (pérdida del HBeAg y aparición del anticuerpo anti-HBeAg) en un 16–18% de los casos al cabo de un año de tratamiento20–22. En los pacientes que no lograron la seroconversión al cabo de este período, se observó un incremento de esta tras la prolongación del tratamiento, que llegó a ser del 27, del 40 y del 47% tras 223, 324 y 4 años25 de monoterapia con lamivudina, respectivamente. Aun en ausencia de seroconversión, el uso continuado de lamivudina puede conseguir por lo menos la reducción de la carga vírica del VHB. La durabilidad de la respuesta una vez suspendido el tratamiento se sitúa en un 77% según un estudio en pacientes de países no asiáticos a los que se siguió durante una media de 36,6 meses26. Por otro lado, en pacientes asiáticos se han obtenido datos menos favorables (alrededor del 50%), probablemente debido al establecimiento de tratamientos más cortos con lamivudina (8–9 meses)27. Los factores que se relacionan con una mayor durabilidad de la seroconversión son edad igual o inferior a 36 años, infección por el genotipo B y tratamiento adicional con lamivudina a partir de la seroconversión igual o superior a 8 meses28. Aproximadamente en la mitad de los pacientes tratados con lamivudina se produce una mejoría histológica y se reduce el riesgo de progresión a fibrosis. La suspensión de la lamivudina puede producir una reagudización transitoria de la hepatitis con aumento de las aminotransferasas. Esto de debe a que el virus se vuelve a replicar a partir de ccADN, una forma críptica de ADN del VHB presente en los hepatocitos de la mayoría de los pacientes con hepatitis B crónica y sobre la que casi no actúan los fármacos antivíricos ni la propia respuesta inmunitaria del huésped. Sin embargo, hay estudios que afirman que la reagudización de la enfermedad ocurre en la misma proporción en los pacientes que suspenden el tratamiento y en los que lo continúan29,30.
Los pacientes con hepatitis B crónica con HBeAg-negativo también presentan altas tasas de reducción de la carga vírica del VHB hasta niveles indetectables dentro del primer año de tratamiento con lamivudina (del 60–70%)31. Sin embargo, alrededor del 90% de los pacientes recae tras la suspensión del fármaco32. Al alargar la duración del tratamiento, se observó un aumento significativo de resistencia a la lamivudina acompañada de una reducción de la respuesta.
La aparición de mutantes resistentes (YMDD) es la principal desventaja de la lamivudina. Se ha descrito en un 14–32% de los pacientes que han recibido lamivudina durante un año, en un 38% de los que la recibieron durante 2 años y en un 66% de los que la recibieron durante 5 años. La presencia de cepas resistentes suele comportar la pérdida de respuesta al tratamiento y puede provocar reagudización de la enfermedad así como descompensación de la hepatopatía en pacientes trasplantados. En los pacientes con trasplante ortotópico de hígado, la dosis de 100mg/día iniciada 4 semanas antes y hasta 90 semanas después parece reducir el riesgo de reinfección. En estos pacientes, el uso de gammaglobulina hiperinmunitaria antihepatitis B, administrada a dosis altas y durante un tiempo indefinido, demostró prevenir la reinfección del injerto si el paciente presentaba una baja replicación vírica en el momento del trasplante, pero no en otros casos.
La lamivudina suele ser un fármaco muy bien tolerado. Puede producir cefalea, náuseas y mareos, aunque no es frecuente. En los niños se han descrito casos de pancreatitis.
AdefovirEl adefovir es un nucleótido monofosfato sintético que se administra como profármaco, el diéster adefovir dipivoxilo, para mejorar su absorción. Requiere tan solo 2 fosforilaciones para su activación y presenta una potente actividad in vitro frente los herpesvirus, los retrovirus y los hepadnavirus. El adefovir actúa inhibiendo la ADN polimerasa vírica y es activo tanto frente a la cepa salvaje del VHB como a las cepas resistentes a lamivudina. En los ensayos de fase iii con pacientes con HBeAg positivo se consiguió la respuesta histológica en el 53% de los pacientes tratados con 10mg al día durante 48 semanas frente al 25% del grupo placebo. La negativización del ADN vírico (ADN del VHB <400 copias/ml) se consiguió en el 21% de los pacientes tratados. La seroconversión se logró en el 14% de los pacientes del grupo experimental en comparación con el 6% de los pacientes que recibieron placebo33. En los pacientes con HBeAg negativo también se logró una buena tasa de respuesta histológica en el grupo tratado con adefovir (10mg/día durante 48 semanas), que fue del 64 frente al 33% del grupo placebo. Además, la respuesta virológica (ADN del VHB <400 copias/ml) fue del 55% en los pacientes que recibieron adefovir34. No se detectaron resistencias a adefovir durante las 48 semanas de tratamiento de los ensayos de fase iii. Sin embargo, se ha observado que al prolongar el tratamiento más allá de un año, aumenta progresivamente la probabilidad de aparición de cepas resistentes hasta cifras del 29% en 5 años35. En presencia de resistencia a lamivudina, se ha demostrado que el riesgo de resistencia a adefovir es mayor cuando se sustituye lamivudina por adefovir que cuando se añade adefovir al tratamiento en curso con lamivudina. La adición del adefovir ha logrado mantener la respuesta al menos durante los 2–3 años de duración de los estudios36,37.
Por otro lado, la asociación de lamivudina con adefovir no ha producido beneficios adicionales respecto a monoterapia con adefovir y cabe destacar que ambas pautas han mostrado una clara superioridad respecto a la monoterapia con lamivudina. En pacientes con coinfección por el VIH y el VHB resistente a lamivudina, la adición de adefovir a lamivudina ha obtenido resultados más favorables que el cambio a adefovir en monoterapia38,39.
A la dosis utilizada (10mg/día), el perfil de seguridad del adefovir es muy bueno. Se ha descrito toxicidad renal en el 8% de los pacientes tratados durante 5 años40 y en el 12% de los pacientes trasplantados, la mayoría de los cuales reciben de forma concomitante fármacos nefrotóxicos como ciclosporina o tacrolimus41. Por esto es recomendable realizar controles frecuentes de la función renal en estos pacientes así como en los casos de cirrosis descompensada.
EntecavirEl entecavir es un nucleósido análogo de la guanosina que actúa inhibiendo la ADN polimerasa vírica por competición con el sustrato natural desoxiguanosina. En los estudios de fase iii, el entecavir demostró de forma significativa una mayor respuesta virológica, histológica y bioquímica que la lamivudina. El estudio realizado con pacientes con HBeAg positivo consiguió a las 48 semanas respuesta histológica en el 72% de los pacientes tratados con entecavir frente al 62% de los pacientes que recibieron lamivudina (p=0,009). Los niveles de ADN del virus fueron indetectables en el 67% de casos del grupo experimental frente al 36% del grupo tratado con lamivudina (p<0,001). Sin embargo, la tasa de seroconversión fue similar en ambos grupos (el 21 frente al 18%) y la pérdida del antígeno de superficie del VHB fue aun menor (el 2 frente al 1%)42. En el estudio de fase iii realizado con pacientes con HBeAg negativo se observó una mayor respuesta histológica en el grupo que recibió entecavir durante 48 semanas respecto al que recibió lamivudina durante el mismo período (el 70 frente al 61%) (p<0,01). La negativización de los niveles de ADN vírico se consiguió en el 90% de los casos tratados con entecavir vs. 72% de los tratados con lamivudina (p<0,001)43.
En los estudios citados anteriormente se evaluó la respuesta sostenida al cabo de 24 semanas de la interrupción del tratamiento en los pacientes con respuesta definida por el protocolo (pérdida del HBeAg y el ADN vírico <0,7mEq/ml para los pacientes HBeAg positivo, y ADN vírico <0,7mEq/ml y ALT <1,25ULN para los pacientes con HBeAg negativo). En los pacientes con HBeAg positivo, la respuesta virológica sostenida (RVS) fue del 82% con entecavir frente al 73% con lamivudina. En cambio, en los pacientes con HBeAg negativo, la respuesta fue claramente menor: el 48% con entecavir y el 35% con lamivudina.
En caso de fracaso de tratamiento con lamivudina en pacientes con HBeAg positivo, los pacientes que cambiaron a entecavir obtuvieron respuestas histológicas, virológicas y bioquímicas superiores a las de los pacientes que continuaron recibiendo lamivudina (el 55, el 21 y el 75% frente al 28, el 1 y el 23%, respectivamente), aunque estas fueron inferiores a las obtenidas en pacientes naïve44.
El entecavir presenta la ventaja de poseer una elevada barrera genética. En pacientes que no hayan recibido análogos de nucleósidos, el desarrollo de resistencia a entecavir es muy poco probable. En los ensayos clínicos de fase iii se ha descrito la reactivación de la replicación vírica en solo el 3% de los pacientes al cabo de 96 semanas de tratamiento45. Sin embargo, el tratamiento previo con lamivudina favorece la aparición de resistencia a entecavir. En un estudio de seguimiento de 4 años de tratamiento con entecavir se estima que la probabilidad de reactivación de la replicación vírica tras el desarrollo de resistencia en pacientes sin tratamiento previo con lamivudina es del 0,8%, mientras que en los pacientes que han presentado fracaso a la lamivudina asciende hasta el 39,5%46.
Contrariamente a lo esperado, el entecavir presenta actividad parcialmente inhibidora de la replicación del VIH-1. Además posee la capacidad de seleccionar la mutación M184V que confiere resistencia de alto nivel a lamivudina, emtricitabina y entecavir. En referencia a los pacientes coinfectados por el VIH y el VHB, se recomienda no utilizar entecavir mientras no reciban tratamiento antirretrovírico concomitante47.
La dosis indicada de entecavir es de 0,5mg una vez al día por vía oral para pacientes sin tratamiento previo con análogos de nucleósidos, mientras que se recomienda 1mg una vez al día en caso de fracaso de tratamiento con lamivudina o telbivudina.
Según los resultados de los ensayos clínicos, el entecavir es un fármaco con un buen perfil de seguridad, y su tolerancia es similar a la de la lamivudina. Aun así, cabe mencionar la reciente publicación de 5 casos de acidosis láctica grave en una serie de 16 casos de pacientes con hepatitis B y cirrosis. La incidencia de acidosis láctica se correlacionó con el índice MELD (Model for End-stage Liver Disease) de tal modo que los pacientes afectados tenían una puntuación igual a 20 o superior, mientras que el resto de pacientes tenía una puntuación inferior a 18 (p<0,005). Los autores destacan la necesidad de evaluar la seguridad del entecavir en estos pacientes en estudios bien diseñados y recomiendan utilizarlo con precaución48. Finalmente, en estudios en animales se ha descrito el desarrollo de neoplasias a dosis de entre 3–40 veces las utilizadas en humanos.
TelbivudinaLa telbivudina es un análogo nucleósido sintético de la timidina que, una vez fosforilada, inhibe la ADN polimerasa del VHB por competencia con el sustrato natural timidina-5′-trifosfato. Se realizó un ensayo de fase iii en 921 pacientes con HBeAg positivo y 446 pacientes con HBeAg negativo, a los que se aleatorizó para recibir tratamiento con telbivudina o lamivudina durante 104 semanas. La respuesta terapéutica se estableció en ADN del VHB <5 log10 copias/ml, negativización del HBeAg o normalización de los niveles de ALT. La telbivudina presentó un porcentaje de respuesta significativamente superior a la lamivudina tanto en pacientes con HBeAg positivo (el 63 frente al 48%) como en pacientes con HBeAg negativo (el 78 frente al 66%). Sin embargo, no se obtuvieron diferencias estadísticamente significativas en la negativización del HBeAg al cabo de uno y 2 años de tratamiento.
La reactivación de la actividad vírica debido al desarrollo de resistencia a telbivudina al cabo del primer año de tratamiento fue del 5% en los pacientes con HBeAg positivo y del 2,3% en los pacientes con HBeAg negativo49. A las 104 semanas, la tasa de resistencia se elevó al 21,6 y el 8%, respectivamente, que fue inferior a la de la lamivudina. Según los estudios realizados, los pacientes tratados con telbivudina tienen mayor riesgo de desarrollar resistencias cuando la carga vírica sigue siendo superior a las 300 copias/ml al cabo de 24 semanas de tratamiento.
En general, las guías de tratamiento del VIH recomiendan iniciar tratamiento antirretrovírico a la vez que el tratamiento del VHB en los pacientes coinfectados. En el caso de que algún paciente con coinfección no deba recibir tratamiento antirretrovírico y se lo deba tratar por el VHB, una de las opciones terapéuticas que se recomiendan en las guías de tratamiento es la asociación de telbivudina y adefovir. En estudios in vitro parece ser que la asociación no tiene actividad sobre el VIH. Sin embargo, se ha descrito un caso de reducción de la carga vírica del VIH con telbivudina más adefovir en ausencia de tratamiento antirretrovírico concomitante. Por tanto, no se puede descartar que la telbivudina presente actividad sobre el VIH39,50,51.
La dosis indicada de telbivudina es de 600mg una vez al día por vía oral y debe reducirse en caso de insuficiencia renal. El perfil de seguridad fue similar a la lamivudina en los ensayos clínicos de fase iii, pero se han observado elevaciones de la creatina fosfocinasa (>7 veces el límite superior de normalidad) y se han descrito casos de miopatía que han remitido con la retirada del tratamiento.
TenofovirEl tenofovir disoproxil fumarato es el profármaco del tenofovir, un análogo de nuleótido que se registró para el tratamiento de la infección por el VIH y que recientemente se ha aprobado para el tratamiento de la hepatitis B crónica, tanto en pacientes coinfectados por el VIH y el VHB como en pacientes infectados por el VHB. Los 2 estudios de fase iii que se han llevado a cabo para la aprobación de la nueva indicación comparan tenofovir 300mg al día con adefovir 10mg al día en pacientes con HBeAg positivo y negativo, sin infección por el VIH. La duración de la fase de tratamiento doble ciego fue de 48 semanas, seguida de una fase de extensión de tratamiento abierto con tenofovir de 240 semanas, actualmente en curso. La eficacia virológica (ADN del VHB <400 copias/ml) fue significativamente superior en los pacientes tratados con tenofovir al cabo de 48 semanas (el 76 frente al 13% en pacientes con HBeAg positivo y el 93 frente al 63% en pacientes con HBeAg negativo). No obstante, no se encontraron diferencias significativas en cuanto a eficacia histológica entre ambos grupos, probablemente porque se requieran períodos más prolongados de tratamiento para que la supresión de la carga vírica se traduzca en un beneficio claro en la progresión de la enfermedad.
En relación con la aparición de resistencias, no se han encontrado alteraciones en la polimerasa vírica que confieran resistencia a tenofovir, aunque en los estudios hubo algunos casos de aumento de la carga vírica que se atribuyeron a un mal cumplimiento. Los pacientes incluidos en el protocolo se seguirán durante 7 años para analizar el desarrollo de resistencia al fármaco. Las elevadas dosis de tenofovir indicadas en el tratamiento de la hepatitis B (300mg respecto a los 10mg de adefovir) podrían explicar la mayor potencia antivírica del tenofovir respecto al adefovir y la alta barrera genética que posee. Estas ventajas convierten al tenofovir en una de las mejores opciones terapéuticas en el tratamiento de la hepatitis B crónica junto con el entecavir.
En caso de coinfección por el VIH y el VHB, se considera que la asociación de tenofovir y emtricitabina o lamivudina es una de las mejores alternativas terapéuticas cuando se cumplen criterios para tratamiento de ambas infecciones víricas52. En caso de que el VIH desarrolle resistencia a tenofovir, puede mantenerse el tratamiento asociado a la nueva pauta antirretrovírica o bien cambiar a entecavir, telbivudina o adefovir39.
El tenofovir es un fármaco bien tolerado, aunque puede provocar insuficiencia renal y se han descrito casos de síndrome de Fanconi. Se recomienda monitorizar la función renal de los pacientes, sobre todo cuando el aclaramiento de creatinina es inferior a 50ml/min, y ajustar la dosis del fármaco.
EmtricitabinaLa emtricitabina es un análogo sintético del nucleósido citidina con actividad sobre el VIH y el VHB. En un estudio realizado en pacientes monoinfectados con el VHB, la emtricitabina fue más eficaz que el placebo. Sin embargo, las tasas de resistencia al cabo de 2 años de tratamiento fueron del 13%53.
Actualmente, la emtricitabina se utiliza para el tratamiento de pacientes coinfectados por el VIH y el VHB en combinación con tenofovir para reducir la posibilidad de desarrollar resistencias al tratamiento, sobre todo en casos de alta carga vírica del VHB. La doble terapia también puede ser beneficiosa en pacientes cirróticos en los que se inicia el tratamiento anti-VIH/VHB para evitar el agravamiento de la hepatitis en el contexto de la reconstitución del sistema inmunitario.
ClevudinaLa clevudina es un nuevo análogo de nucleósido de la pirimidina que actúa inhibiendo la ADN polimerasa vírica. Según estudios preclínicos, además de inhibir la síntesis de ADN del virus en el suero, la clevudina presenta actividad en la reducción del ccADN que se sintetiza en los hepatocitos54. Actualmente, la clevudina es un producto en investigación y se están completando los ensayos clínicos de fase iii. En estudios de búsqueda de dosis, la clevudina fue bien tolerada durante períodos de 12 semanas y se estableció la pauta de 30mg una vez al día como pauta óptima. Tras un período de tratamiento de 24 semanas, se obtuvo respuesta virológica (ADN del VHB <300 copias/ml) en el 59% de los pacientes con HBeAg positivo y en el 92,1% de los pacientes con HBeAg negativo. La respuesta bioquímica (normalización de los valores de ALT) se obtuvo en el 68,2 y el 74,6%, respectivamente. Al cabo de 24 semanas de la retirada del tratamiento, el descenso de los niveles de ADN del virus fue de 5,10–2,02 log10 copias/ml en los pacientes con HBeAg positivo y de 4,25 a 3,11 log10 copias/ml en los pacientes con HBeAg negativo. De estos últimos, solo el 16,1% continuó presentando carga vírica indetectable.
Recientemente, en pacientes tratados con clevudina, se ha descrito la aparición de miopatía causada por depleción del ADN mitocondrial55.
Nuevos fármacosSe encuentran en investigación diversos fármacos para el tratamiento de la hepatitis B, de entre los que destacan la timosina α-1 (un polipéptido con actividad inmunomoduladora), la nitazoxanida, utilizada como antiparasitario, y sus análogos56, y el celgosivir, un inhibidor de la glicosidasa, actualmente en fase ii. Además, en fase preclínica se encuentran nuevas clases de antivíricos como las heteroalildihidropirimidinas, que impiden la formación de la nucleocápside del virus; las fenilpropenamidas, que inhiben la encapsidación, y los análogos de la helioxantina, que actúan inhibiendo la expresión del ADN y el ARN del VHB. Se está investigando también en vacunas terapéuticas para estimular la inmunidad celular frente a los antígenos del VHB, aunque de momento se trata de estudios de fases i y ii57.
Antivíricos activos frente al virus de la hepatitis CLa infección crónica por el VHC es la causa más frecuente de enfermedad crónica del hígado y la primera causa de trasplante hepático. Su tendencia a la remisión espontánea es escasa y su progresión suele ser lenta. Los signos y los síntomas de fallo hepático aparecen cuando la enfermedad está ya muy avanzada. Se cree que alrededor del 20% de los pacientes pueden desarrollar cirrosis después de los 30 años de enfermedad. Además, la infección crónica por el VHC es un factor de riesgo para el desarrollo de carcinoma hepatocelular, cuya tasa de incidencia anual oscila entre el 1-4% de los pacientes infectados. El pronóstico a largo plazo es difícil de establecer, en muchos pacientes la enfermedad se mantiene estable durante un largo período de tiempo, en cambio, en otros, puede progresar de forma rápida. Algunos factores pueden comportar un peor pronóstico, como son el consumo habitual de alcohol (incluso moderado) y de cannabis, el sexo masculino, la adquisición de la infección por encima de los 40 años y la función deficiente del sistema inmunitario (p. ej., infección por el VIH).
La decisión de tratar a un paciente con hepatitis C crónica suele ser difícil y a menudo se basa en los hallazgos de la biopsia hepática. Los pacientes que más pueden beneficiarse del tratamiento son los que presentan una enfermedad moderada y los que presentan infección por alguno de los genotipos favorables (2 y 3). Sin embargo, aunque el potencial beneficio sea menor, también deben valorarse para tratamiento aquellos casos que han progresado a cirrosis, los pacientes alcohólicos, los que presentan coinfección por el VIH y los drogadictos por vía parenteral.
El tratamiento de elección es el PEG-IFN combinado con ribavirina (RBV), tanto en pacientes naïve como en los que han tenido recaídas después de responder al IFN en monoterapia. La combinación obtiene mejores resultados y, adicionalmente, el PEG-IFN (conjugado con una o varias moléculas de polietilenglicol) obtiene mejores resultados en comparación con el IFN clásico. El principal objetivo del tratamiento es alcanzar la RVS, es decir, que el ARN del virus sea indetectable por PCR de forma mantenida durante los 6 meses posteriores a la finalización del tratamiento. Para esto, es imprescindible una adecuada adherencia al tratamiento.
El factor pronóstico de respuesta más importante en el tratamiento del VHC es el genotipo. En los pacientes con genotipo 1, el porcentaje de respuesta al tratamiento con PEG-IFN en combinación con RBV es del 40–45% comparado con aproximadamente el 80% de los que tienen genotipo 2 o 3. Entre otros factores de buen pronóstico se encuentran la ausencia de cirrosis, la carga vírica inicial baja, el sexo femenino y una edad inferior a 40 años. La duración del tratamiento se suele establecer en función del genotipo. En los genotipos 2 o 3, la respuesta terapéutica se consigue con 24 semanas de tratamiento con dosis bajas de RBV (800mg/día). En cambio, en la infección por el genotipo 1, se precisan 48 semanas de tratamiento y son necesarias dosis mayores de RBV ajustadas al peso del paciente (800mg si el peso es inferior a 65kg, 1.000mg si el peso está entre 65–85kg, 1.200mg si el peso está entre 85–105kg, y 1.400mg si el peso es superior a 105kg pero inferior a 125kg). Si no se ha obtenido respuesta a las 24 semanas, el tratamiento debe suspenderse, ya que una respuesta futura es poco probable. En los paciente con genotipo 1 se puede predecir la consecución de la RVS en función de la respuesta virológica a las 12 semanas. Del 97–100% de estos pacientes serán no respondedores y, por tanto, se recomienda la retirada del tratamiento. Sin embargo, hay estudios que indican que la prolongación del tratamiento a 72 semanas puede ser beneficiosa en pacientes con genotipo 1, en los que la negativización de la PCR se obtiene entre las 12–24 semanas de tratamiento58.
El riesgo de infección por el VHC es especialmente elevado en los pacientes que reciben hemodiálisis y aumenta en función del tiempo. En estos pacientes es preferible la monoterapia con IFN estándar durante 12 meses, independientemente del genotipo. También puede utilizarse una dosis menor de PEG-IFN, aunque aumenta la probabilidad de efectos adversos en comparación con el IFN estándar. Finalmente, se puede asociar RBV al tratamiento a dosis de 200mg al día, monitorizando estrechamente la aparición de reacciones adversas. Si a los 3 meses de tratamiento la PCR sigue positiva y las aminotransferasas no mejoran, el tratamiento puede suspenderse, ya que una respuesta posterior es poco probable. Aunque inicialmente un 50% de los pacientes responde a la monoterapia con IFN, tan solo un 3–19% mantiene después una respuesta sostenida. Con monoterapia que utiliza PEG-IFN-α, la respuesta sostenida es mayor, alrededor del 25%. Actualmente se recomienda realizar tratamiento anti-VHC antes del trasplante renal en los pacientes candidatos dada la peor supervivencia en trasplantados con infección por el VHC.
En los pacientes con cirrosis compensada se recomienda realizar el tratamiento estándar de PEG-IFN y RBV, vigilando atentamente el desarrollo de reacciones adversas, que suelen ser más frecuentes en esta población. En los pacientes con cirrosis descompensada, es altamente recomendable erradicar el VHC antes del trasplante dado el alto riesgo de reinfección del injerto si no se elimina el virus. En estos pacientes, el tratamiento debe iniciarse bajo estrecho control médico, con dosis reducidas de PEG-IFN y RBV, Los pacientes infectados por los genotipos 2 y 3 presentan mayor probabilidad de respuesta que los del genotipo 158.
En los países desarrollados, alrededor del 25% de los pacientes infectados por el VIH presenta coinfección por el VHC. En estos pacientes es altamente importante instaurar el tratamiento para el VHC, ya que el curso de la enfermedad hepática suele ser más rápido y el riesgo de cirrosis se duplica. Además, la probabilidad de obtener la RVS en estos pacientes es menor. Se recomienda seguir el mismo tratamiento que los pacientes monoinfectados por el VHC con PEG-IFN y RBV durante 48 semanas, independientemente del genotipo. Las guías indican que se podría considerar reducir la duración a 24 semanas en los genotipos 2 y 3 cuando la viremia se negativiza a la semana 439. En un metaanálisis recientemente publicado, las tasas de RVS fueron del 26% para los genotipos 1 y 4 y del 57% para los genotipos 2 y 359.
Finalmente, recientemente se han publicado dos estudios de asociación del genoma humano (genome-wide association)60,61 que han encontrado un polimorfismo de un solo nucleótido (SNP) próximo al gen de la IL28B que codifica la producción de interferon-lambda () y que se ha asociado al aclaramiento espontáneo de la infección por el VHC en pacientes VIH negativos (de ahí que las tasas de aclaramiento sean distintas según las razas, mayor en población asiática y menor en la de ascendencia africana) y en la tasa de respuesta virológica sostenida al tratamiento con interferón-pegilado y ribavirina, que es como mínimo dos veces mayor en aquellos pacientes infectados por el VHC tanto VIH negativos62 como VIH positivos63 que tenían un genotipo de la IL28B silvestre. En un estudio suizo, este efecto beneficioso solo se observó en pacientes con genotipo 1 o 4. Resultados similares se han visto en pacientes coinfectados por el VIH y el VHC4. Es muy probable que dada la trascendencia de este hallazgo la determinación de los polimorfimos de la IL28B se pueda incorporar a corto-medio plazo en la práctica clínica.
InterferónExisten 2 tipos de IFN-α comercializados en España para el tratamiento de la hepatitis C crónica: el IFN-α-2a y el IFN-α-2b, tanto en su forma estándar como pegilada. Todas las formas de IFN-α parecen ser similares, en cuanto a eficacia, frente a la hepatitis C. Como ya se ha comentado anteriormente, se utilizan en combinación con RBV, ya que la asociación se ha relacionado con tasas más altas de respuesta que con cualquiera de los 2 fármacos en monoterapia. En los pacientes tratados con IFN se ha demostrado una reducción de la incidencia de carcinoma hepatocelular. Sin embargo, en algunos casos se ha descrito la aparición de anticuerpos durante el tratamiento con IFN, con reducción de su eficacia.
El IFN-α-2a (Roferon A®) se administra a una dosis de 3-4,5MU 3 veces por semana por vía subcutánea o intramuscular durante 6–12 meses en combinación con RBV. En pacientes que no toleren la RBV o en caso de que esta esté contraindicada, se administra monoterapia con IFN-α-2a a una dosis inicial de 3-6MU 3 veces por semana durante 6 meses, seguida de 3MU 3 veces por semana 6 meses adicionales o, como alternativa, 3MU 3 veces por semana durante 12 meses, por vía subcutánea o intramuscular.
El IFN-α-2b (Intron A®) se administra a dosis de 3MU 3 veces por semana durante 6–12 meses en combinación con RBV o, en caso de que esta no pueda utilizarse, como monoterapia durante 12–18 meses y hasta 24 meses, por vía subcutánea o intramuscular.
Actualmente se encuentran comercializadas en España las formas pegiladas de IFN-α-2b e IFN-α-2a, con las que se consigue retrasar la eliminación y aumentar el área bajo la curva de concentraciones plasmáticas de IFN. El PEG-IFN-α-2b está formado por la unión covalente de una molécula de monometoxipolietilenglicol de 12kDa a una molécula de IFN-α-2b y el PEG-IFN-α-2a está formado por la unión covalente a una molécula de monometoxipolietilenglicol de 40kDa a una molécula de IFN-α-2a. Con la unión de monometoxipolietilenglicol se consigue aumentar el tamaño molecular del fármaco, lo que reduce la filtración glomerular y aumenta considerablemente su semivida de eliminación: de 7–9h–40h para PEG-IFN-α-2b y de 6–9h a 72–96h para PEG-IFN-α-2a. Las concentraciones séricas máximas con una administración semanal de PEG-IFN son superiores a las conseguidas con 3 inyecciones semanales de IFN estándar.
El PEG-IFN-α-2b (PegIntron®) se administra a una dosis de 1–1,5mcg/kg una vez por semana durante 6–12 meses en combinación con RBV o bien, cuando no es posible utilizar esta, de 1mcg/kg una vez por semana durante 12 meses como monoterapia.
El PEG-IFN-α-2a (Pegasys®) se administra a una dosis de 180 mcg semanales durante 6–12 meses en combinación con RBV o bien, cuando no es posible utilizar esta, de 180 mcg semanales durante 12 meses como monoterapia.
Con cualquiera de las 2 nuevas formas pegiladas de IFN-α, la eficacia es superior a la presentación clásica, tanto en monoterapia (10–39% comparado con 3–19%) como en combinación con RBV. En pacientes con genotipo 1, la RVS es del 42% con PEG-IFN+RBV comparado con el 33% con IFN clásico+RBV.
El uso del IFN está contraindicado en el embarazo y la lactancia, en caso de cardiopatía inestable o no controlada e historia de trastorno psiquiátrico grave, especialmente depresión. Debe administrarse con precaución en pacientes con enfermedades autoinmunitarias, alteraciones tiroideas, diabetes, mielosupresión, neumopatía obstructiva crónica, embolia pulmonar, alteraciones neurológicas (p. ej., epilepsia), retinopatía, enfermedad cardiovascular, insuficiencia renal crónica o cirrosis moderada-grave. Algunos pacientes con psoriasis han referido exacerbaciones durante el tratamiento con IFN. En pacientes con trombocitopenia no debe utilizarse la vía intramuscular.
Durante el tratamiento con IFN-α debe mantenerse una hidratación adecuada. Es necesario realizar controles hematológicos periódicos, especialmente en pacientes con riesgo de mielosupresión, y ajustar la dosis en caso necesario. En pacientes predispuestos a retinopatía, se recomienda realizar un examen oftalmológico previo al inicio del tratamiento. También se recomienda examinar la función cardíaca. El tratamiento debe interrumpirse en pacientes con hepatitis crónica que desarrollen descompensaciones mientras lo estén recibiendo. Por otro lado, debe advertirse a los pacientes que experimenten fatiga, somnolencia o confusión con IFN que eviten la conducción y el manejo de maquinaria peligrosa.
El IFN-α es un fármaco mal tolerado. Hasta un 15% de los pacientes deben abandonar el tratamiento. La inyección subcutánea de IFN se asocia frecuentemente a un síndrome gripal con fiebre, escalofríos, dolor de cabeza y artromialgias, especialmente durante la primera semana de tratamiento. Estos síntomas están relacionados con la dosis y pueden aliviarse con paracetamol. Otros efectos adversos que pueden obligar a la suspensión del tratamiento o a una reducción en la dosis son mielosupresión, astenia intensa, pérdida de peso, aumento de las infecciones bacterianas y reacciones psiquiátricas (depresión [hasta en un 25% de los casos], ansiedad, labilidad emocional y agitación). Pueden producirse también náuseas, vómitos, alteración en las pruebas de función hepática, hipotensión o hipertensión, alopecia, alteraciones tiroideas (hipotiroidismo o hipertiroidismo), acúfenos, hipoacusia reversible, retinopatía, formación de autoanticuerpos y, posiblemente, cardiotoxicidad. Se han descrito casos de insuficiencia renal y síndrome nefrótico, edema pulmonar y neumonitis, agravamiento de diabetes de tipo 2, alteraciones neurológicas (somnolencia, ataxia, parestesias, confusión y, raramente, convulsiones y coma), así como alteraciones visuales y, raramente, retinopatía isquémica. A dosis altas puede causar hipocalcemia y otras alteraciones electrolíticas. Las reacciones en el punto de inyección, neutropenia y anemia relacionadas con la dosis han sido más frecuentes con PEG-IFN. Los factores estimulantes de colonias de granulocitos pueden tener utilidad en caso de neutropenia grave en pacientes con cirrosis avanzada. La depresión causada por el IFN-α se puede tratar con un antidepresivo inhibidor de la recaptación de la serotonina, sin que sea necesario retirar el fármaco. El tratamiento con IFN puede desencadenar o empeorar la hepatitis autoinmunitaria crónica y otras enfermedades autoinmunitarias como la tiroiditis.
RibavirinaLa RBV es un análogo de nucleósidos sintético estructuralmente relacionado con la guanina. Se fosforila a través de enzimas celulares y se piensa que el monofosfato y el trifosfato son los encargados de su actividad. Su mecanismo de actuación es todavía desconocido, aunque actúa interfiriendo en la síntesis del ácido nucleico vírico. En clínica se utiliza por vía oral en el tratamiento de la infección por el VHC, siempre en combinación con IFN-α, ya que la RBV sola no ha mostrado beneficio alguno. Probablemente esto se deba a que no actúa directamente frente al virus, sino modulando el sistema inmunitario para mejorar la respuesta T-helper. Por vía inhalada, la RBV se utiliza en el tratamiento del virus respiratorio sincitial. También se ha utilizado en infecciones graves por adenovirus y en algunos casos de fiebre hemorrágica, incluyendo la fiebre de Lassa, y ha reducido la mortalidad. No se han observado resistencias a RBV, probablemente debido a sus múltiples mecanismos de acción.
La utilización de RBV en asociación con IFN-α en el tratamiento de la hepatitis C ha dado lugar a tasas de respuesta mantenida superiores a las conseguidas con las respectivas monoterapias. Los pacientes que recidivan tras monoterapia con IFN pueden responder a la asociación. Existen 2 preparados comerciales de RBV: Rebetol® cápsulas de 200mg y Copegus® comprimidos con cubierta pelicular de 200mg, que deben utilizarse, respectivamente, en combinación con IFN-α-2b e IFN-α-2a. La RBV debe administrarse junto con alimentos y se dosifica en función del peso corporal y del preparado comercial. Para Rebetol® las dosis son de 400mg en el desayuno y 400mg en la cena para pacientes con menos de 65kg, de 400mg en el desayuno y 600mg en la cena para pacientes de 65–85kg, de 600mg en el desayuno y 600mg en la cena para pacientes de 85–105kg, y de 600mg en el desayuno y 800mg en la cena para pacientes con más de 105kg. Para Copegus® las dosis son de 400mg en el desayuno y 600mg en la cena para pacientes de 75kg o menos, y de 600mg en el desayuno y en la cena para pacientes de más de 75kg. Los comprimidos no se deben romper ni triturar. Algunos autores recomiendan utilizar dosis menores (800mg/día) para pacientes con genotipos 2 y 3, siempre a criterio del médico y en función del peso.
Entre sus principales efectos secundarios destacan la anemia por hemólisis y la mielosupresión. También es frecuente la aparición de fatiga y depresión, problemas que tiene en común con el IFN.
Es necesario realizar una analítica de control previa al inicio del tratamiento, tras 2 y 4 semanas de iniciarlo y después periódicamente, y ajustar la dosis de IFN/RBV en caso de toxicidad. En los pacientes que desarrollen mielotoxicidad debe considerarse la utilización de factores de crecimiento como eritropoyetina, darbepoetina o estimulantes de colonias de granulocitos.
El tratamiento debe mantenerse un mínimo de 6 meses en combinación con IFN-α. En los pacientes con genotipo 1 y negativización del ARN del VHC a los 6 meses, el médico especialista deberá ser quien tome la decisión de prolongar el tratamiento durante un año sobre la base de otros factores, p. ej., carga vírica basal elevada.
La ribavirina es teratógena y embriotóxica en animales, por lo que el embarazo está contraindicado tanto cuando es la mujer la que recibe el tratamiento (hasta trascurridos 4 meses después de su finalización) como si es su pareja la que lo recibe, ya que la RBV puede detectarse en el esperma (en este caso debe esperarse hasta trascurridos 7 meses después de la finalización del tratamiento). Asimismo, las mujeres embarazadas no deben cuidar a pacientes tratados con RBV en aerosol y los hombres en tratamiento cuya pareja esté embarazada deben utilizar preservativo. La ribavirina está también contraindicada durante el período de lactancia, en caso de hepatitis autoinmunitaria o insuficiencia hepática grave, enfermedad psiquiátrica (en especial depresión grave), alteración de la función tiroidea preexistente no controlada, así como en aquellas enfermedades que pudieran verse agravadas por una posible hemólisis inducida por este fármaco, como cardiopatía grave o hemoglobinopatía, ya que la RBV puede producir anemia hemolítica, en algunos casos asociada a hiperbilirrubinemia o hiperuricemia. La RBV oral asociada a IFN parece causar más incidencia de tos, prurito y erupciones cutáneas que el IFN solo. En pacientes predispuestos puede aparecer gota. Otros efectos secundarios descritos han sido anorexia, dispepsia, náuseas, mareo, insomnio, irritabilidad, disnea, faringitis y síndrome seco. En la actualidad se están investigando 2 nuevos fármacos derivados de RBV, levovirina y virmidina, con el objetivo de mantener la actividad inmunomoduladora de la RBV y reducir la toxicidad.
Dado que la RBV se elimina mayoritariamente por vía renal, se recomienda reducir la dosis de mantenimiento en casos de insuficiencia renal y vigilar atentamente la aparición de efectos adversos.
Cuando se decide tratar la infección por el VHC en pacientes coinfectados por el VIH que reciben tratamiento antirretrovírico, deben tenerse en cuenta las interacciones que se han descrito entre la RBV y los diferentes antiretrovíricos. Una de las interacciones descritas se produce con la asociación de RBV con didanosina o estavudina por haberse relacionado con toxicidad mitocondrial. En este caso, se recomienda evitar el tratamiento concomitante. Conviene evitar también la asociación de RBV y zidovudina por el desarrollo de toxicidad hematológica significativa. Por otro lado, hay cierta controversia referente a la posible disminución de la eficacia del tratamiento del VHC (IFN más RBV) cuando se administra con abacavir. Hasta que no haya datos concluyentes se recomienda evitar la asociación en aquellos pacientes con menor probabilidad de respuesta39.
TelaprevirEl telaprevir es un inhibidor específico de la serinproteasa del VHC. Sus efectos adversos más frecuentes son exantema, prurito y anemia. McHutchison et al64 presentaron los resultados del estudio PROVE1, un estudio multicéntrico, aleatorizado y controlado con placebo, en el que se comparó la RVS obtenida con pautas de telaprevir (1.250mg el primer día, seguido de 750mg c/8h) durante 12 semanas, combinado con PEG-IFN-α-2a (180mcg/semana) y RBV (1.000–1.200mg/día en función del peso) 12, 24 y 48 semanas con la RVS obtenida en el grupo control que recibió PEG-IFN+RBV durante 48 semanas. En el grupo control, la tasa de RVS fue del 41% en comparación con el 35, el 61 y el 67%, respectivamente, en los que recibieron PEG-IFN+RBV+telaprevir durante 12, 24 y 48 semanas, respectivamente. El porcentaje de abandonos en los grupos que recibieron telapevir fue del 21% (especialmente por exantema) en comparación con un 11% en el grupo control. Hézode et al65 presentaron los resultados del estudio PROVE2, un estudio de fase 2 en el que se incluyeron 334 pacientes con infección crónica por el VHC y genotipo 1 sin tratamiento previo, a los que se aleatorizó para recibir 4 tratamientos. El grupo control recibió PEG-IFN-α-2a (180mcg/semana) + RBV (dosis ajustada al peso) durante 48 semanas. En este grupo, un 46% de los pacientes alcanzaron una RVS. Los otros 3 grupos realizaron un tratamiento más corto y recibieron adicionalmente telaprevir (1.250mg el primer día, seguido de 750mg c/8h) durante 12 semanas junto con PEG-IFN +/− RBV. En el grupo que no recibió RBV, el porcentaje de RVS fue significativamente inferior (36%). En el grupo que recibió RBV fue del 60% y uno de los grupos, que recibió además PEG-IFN y RBV otras 12 semanas adicionales, obtuvo una RVS del 69%, significativamente superior al grupo control.
Nuevos fármacosNuevos compuestos en fase de investigación incluyen inhibidores específicos de serinproteasas del VHC e inhibidores de la ARN polimerasa, además de otras moléculas. Entre los inhibidores de la proteasa, se encuentran el telaprevir y el boceprevir, actualmente en fase iii. Los ensayos de fase ii en pacientes con el genotipo 1 han obtenido una mayor RVS con la asociación del inhibidor de la proteasa a PEG-IGN y RBV que con PEG-IGN y RBV solos.
Los inhibidores de la polimerasa del VHC se dividen en análogos y no análogos de nucleósidos. Entre los análogos se encuentran la valopicitabina, el R1479, el R1626 y el R7128, entre los no análogos se encuentra el HCV796. Todos ellos están en las fases i y ii de investigación clínica.
El próximo paso será el diseño de estudios para evaluar la eficacia y la seguridad de las combinaciones de inhibidores de la proteasa con inhibidores de la polimerasa, asociados o no a RBV. Deberá establecerse también si estas nuevas estrategias terapéuticas permiten en un futuro prescindir del tratamiento con IFN66.
Antivíricos activos frente al virus de la influenzaLa gripe es una enfermedad causada por un virus ARN. Se distinguen 3 grandes tipos de virus: A, B y C. El virus C es el menos dañino de los 3, y causa únicamente infecciones respiratorias leves que resuelven sin necesidad de tratamiento. Los virus A y B en ocasiones pueden dar lugar a infecciones más graves, y por esto se han desarrollado estrategias terapéuticas de vacunación y quimioprofilaxis. La particularidad del virus de la influenza es su gran facilidad para evadir la respuesta del sistema inmunitario. Esto se debe, por un lado, a la elevada frecuencia de cambios antigénicos rápidos e impredecibles que presenta y, por otro lado, a la aparición de diferentes subtipos por recombinación con virus de la influenza que afectan a otras especies (p. ej., aves y cerdos). Aunque no son efectivas al 100%, tanto la vacunación como la quimioprofilaxis reducen en gran medida la propagación de la enfermedad y su morbimortalidad. La vacunación presenta una efectividad del 75–80% en adultos sanos y alrededor del 40% en ancianos residentes en centros geriátricos. Está recomendada en individuos mayores de 65 años y en aquellos que tienen enfermedades crónicas, así como en el personal sanitario. Disponemos de 2 clases de fármacos antivíricos para tratamiento y quimioprofilaxis: los inhibidores de la fusión (amantadina y rimantadina) y los inhibidores de la neuraminidasa (zanamivir y oseltamivir). Estos fármacos pueden reducir la gravedad y la duración de la enfermedad en 1,5–2,5 días. La efectividad es mayor si se administran en las primeras 48 h desde el inicio de la sintomatología.
El oseltamivir presenta la ventaja de su administración por vía oral con una buena biodisponibilidad (75%) mientras que la rimantadina tiene una eficacia similar a la amantadina pero con mejor tolerancia, ya que cruza con mayor dificultad la barrera hematoencefálica. Sin embargo, la rimantadina no se encuentra comercializada en España.
A continuación se describen los principales antivíricos utilizados en las infecciones por el virus de la influenza, así como los nuevos fármacos en investigación.
AmantadinaLa amantadina es un antivírico de acción virostática que actúa impidiendo el acceso del ARN vírico al interior celular. Es activo frente a virus de la influenza tipo A (H1N1), pero no se recomienda para el tratamiento de la gripe por el virus A (H3N2) por haberse descrito altos niveles de resistencia a partir de 2006. Si se inicia el tratamiento oral tras la exposición, tiene una eficacia preventiva del 70–90%. El tratamiento iniciado en un plazo de 48h desde el inicio del cuadro gripal reduce la duración de la fiebre y los síntomas. Se desconoce si reduce también la incidencia de complicaciones o si es eficaz en la neumonía gripal.
La profilaxis con amantadina se ha utilizado para controlar brotes de gripe en residencias y centros sanitarios y para proteger a los pacientes de alto riesgo vacunados después del inicio de la estación gripal. En este último caso, debe administrarse amantadina tan solo durante las 2 semanas posteriores a la vacunación (6 semanas en niños menores de 9 años que se vacunan por primera vez ya que requieren 2 dosis de vacuna separadas 4 semanas).
La amantadina es rápidamente absorbida en el tracto digestivo. Se elimina principalmente por vía renal. Su semivida de eliminación es de 11–15h, pero puede ser mayor en ancianos y en presencia de insuficiencia renal, por lo que deben reducirse las dosis. La amantadina atraviesa las barreras placentaria y hematoencefálica y se distribuye a la leche materna.
Pueden producirse fallos terapéuticos debidos a la rápida emergencia de virus resistentes (hasta en un 33% de los pacientes durante los primeros 5 días de tratamiento). La amantadina presenta resistencia cruzada con la rimantadina. La resistencia se produce por mutación en un solo nucleótido de la proteína M2, y mantiene la virulencia. Los virus resistentes a amantadina son sensibles a oseltamivir y a zanamivir.
Las dosis indicadas en adultos y niños mayores de 9 años son de 200mg al día, tanto en tratamiento como en profilaxis. En niños de uno a 9 años se recomiendan 5–8mg/kg al día sin sobrepasar los 150mg.
La amantadina está contraindicada en insuficiencia renal grave así como en pacientes con historia de epilepsia o ulcus gástrico. Debe utilizarse con precaución en pacientes con enfermedades cardiovasculares o hepáticas, insuficiencia renal moderada, eccema recurrente o psicosis, así como en pacientes ancianos, ya que estos últimos pueden presentar una mayor susceptibilidad a los efectos antimuscarínicos del fármaco y pueden tener la función renal alterada. No debe administrarse durante el embarazo y la lactancia, ya que ha mostrado embriotoxicidad y teratogenia en animales y se han descrito efectos secundarios en niños amamantados por mujeres en tratamiento con amantadina.
La amantadina puede causar anorexia, náuseas, edema periférico y efectos anticolinérgicos. Especialmente en los ancianos, puede causar efectos leves sobre el SNC (nerviosismo, ansiedad, insomnio, dificultad en la concentración, mareo, dolor de cabeza, pesadillas, cambios de humor), que suelen mejorar tras la primera semana de tratamiento y desaparecen rápidamente cuando éste se interrumpe. Se han producido efectos centrales más graves (confusión, alucinaciones, convulsiones) en pacientes con factores de riesgo: edad avanzada, insuficiencia renal, trastornos convulsivos, tratamiento anticolinérgico concomitante o con enfermedad psiquiátrica subyacente.
La amantadina se utiliza también como antiparkinsoniano. Puede potenciar los efectos secundarios de los fármacos anticolinérgicos y de la levodopa, y debe utilizarse con precaución junto con estimulantes del SNC. Su eliminación renal puede aumentarse acidificando la orina.
ZanamivirEs un inhibidor de la neuraminidasa del virus de la gripe. Esta enzima, que se encuentra ubicada en la superficie del virus, es esencial para la liberación de las nuevas partículas víricas formadas y puede también facilitar el acceso del virus a través de las mucosas. Se utiliza el polvo inhalado por vía oral para el tratamiento de la gripe aguda no complicada por el virus de la influenza tipo A (H1N1), A (H3N2) y el virus tipo B en adultos, adolescentes y niños mayores de 5 años. Como tratamiento se administran 2 inhalaciones (equivalentes a una dosis de 10mg) cada 12h durante 5 días, y como profilaxis se administran 2 inhalaciones una vez al día. Uno de sus principales inconvenientes es la dificultad que plantea la administración inhalada por vía oral en niños y ancianos. Cabe recordar que el polvo para inhalación no debe reconstituirse con ningún líquido ni administrarse a través de nebulización o ventilación mecánica. El laboratorio Glaxo SmithKline y las agencias reguladoras han emitido recientemente un comunicado a los profesionales de la salud en relación con un caso de muerte por empleo de zanamivir a través de un dispositivo de ventilación mecánica, con obstrucción de éste67.
La absorción sistémica del zanamivir inhalado es del 10–20% de la dosis, por tanto, no se considera necesario realizar ajustes en caso de insuficiencia renal. Si se inicia el tratamiento dentro de los 2 días posteriores al inicio de los síntomas, puede acortar la duración de la enfermedad y reducir la incidencia de algunas complicaciones respiratorias.
La aparición de resistencias a zanamivir es poco habitual, mientras que es más frecuente para oseltamivir (hasta el 4% de los casos).
El zanamivir inhalado es generalmente bien tolerado. Los efectos secundarios más habituales son molestias nasales y de la garganta, tos, dolor de cabeza, molestias digestivas, síntomas algunos de los cuales son difíciles de distinguir de los causados por la propia enfermedad. Se han descrito raramente casos de broncoespasmo en pacientes con enfermedad reactiva de las vías respiratorias, que han sido mortales en algunos casos, por lo que debe administrarse con precaución en estos pacientes. Además, es recomendable tener a mano un broncodilatador de acción rápida. También se han descrito algunas reacciones de hipersensibilidad.
OseltamivirEl oseltamivir es también un inhibidor de la neuraminidasa del virus de la gripe. Presenta actividad sobre el virus de la influenza A y B, sin embargo algunas cepas de virus A (H1N1) son resistentes a oseltamivir, aunque permanecen susceptibles a zanamivir y amantadina. El nuevo virus de la gripe A (H1N1) detectado inicialmente en México en abril de 2009 es sensible a este antivírico.
En los ensayos clínicos para la comercialización del fármaco en adultos sanos, el oseltamivir ha demostrado una ligera reducción de alrededor de un día de enfermedad y menores títulos nasales de virus cuando el tratamiento se iniciaba dentro de las 48h siguientes al inicio de los síntomas. En otro estudio en adultos sanos y en adultos con comorbilidad, se obtuvo un menor número de complicaciones respiratorias tributarias de tratamiento antibiótico y una reducción de la hospitalización de los pacientes68.
A diferencia del zanamivir, el oseltamivir se administra por vía oral y se encuentra comercializado en cápsulas y en polvo para suspensión oral. Está indicado en el tratamiento y la profilaxis de la influenza en niños mayores de un año, adolescentes y adultos. En adultos se administran 75mg c/12h durante 5 días para tratamiento y 75mg una vez al día como profilaxis. Basándose en la evidencia científica disponible hasta el momento, y en ausencia de indicación, la EMEA considera que en caso de pandemia, los niños menores de un año y las mujeres embarazadas pueden recibir tratamiento o profilaxis con oseltamivir. Las dosis indicadas en niños menores de un año son de 2–3mg/kg c/12h durante 5 días para tratamiento y 2–3mg/kg una vez al día como profilaxis. En pacientes con insuficiencia renal, se recomienda reducir la dosis de oseltamivir cuando el aclaramiento de creatinina es inferior a 30ml/min. El fabricante no recomienda la administración en casos de aclaramientos inferiores a 10ml/min o en la diálisis. Sin embargo, los autores de un estudio en pacientes con diálisis peritoneal y hemodiálisis, recomiendan 30mg una vez por semana y 30mg cada 2 sesiones posthemodiálisis en cada una de las 2 técnicas respectivamente, tanto para tratamiento como para profilaxis69.
El oseltamivir tiene un buen perfil de seguridad. Como reacciones adversas más frecuentes se encuentran las molestias gastrointestinales (náuseas, vómitos y diarrea) y la cefalea. Se han notificado también algunos casos de hipersensibilidad.
Nuevos fármacosEl peramivir es un nuevo inhibidor de la neuraminidasa en investigación. Tiene una baja biodisponibilidad por vía oral y por eso debe administrarse por vía intravenosa o intramuscular. Presenta actividad frente a los virus de la influenza A y B. In vitro ha demostrado una potencia mayor que el zanamivir o el oseltamivir y las cepas resistentes son menos virulentas. También se encuentran en fase de desarrollo clínico inhibidores inhalados de la neuraminidasa de larga duración, como el CS-8958, que se administra una vez a la semana70.
El T-705 es un fármaco que actúa inhibiendo la ARN polimerasa del virus, y es útil en el tratamiento de la influenza A, B, C y de algunas fiebres hemorrágicas. Finalmente, se están realizando estudios con una nueva molécula (DAS181) que inhibe la unión del virus a los receptores de membrana de la célula70.

