




Introducción
Enterococcus faecalis es un habitante comensal del tracto gastrointestinal humano y de animales, pero desde hace años se conoce también su papel como agente causante de endocarditis infecciosa1 y en la actualidad es un reconocido patógeno nosocomial2. La importancia de E. faecalis desde un punto de vista clínico y epidemiológico se debe principalmente a su capacidad para adquirir elementos genéticos que codifican determinantes de virulencia y resistencia antibiótica que pueden mejorar su adaptación a ecosistemas complejos como es el hospitalario3. El paradigma de esta evolución y adaptación ha sido la aparición y diseminación de cepas de enterococos resistentes a vancomicina4.
Recientemente se ha definido la estructura poblacional de E. faecium5,6 y E. faecalis7 empleando la técnica de Multilocus sequence typing (MLST). Como ha sido demostrado, MLST es la técnica más adecuada para el estudio de la estructura poblacional y los mecanismos implicados en la generación de diversidad genética en las poblaciones bacterianas8,9. Además, el empleo de la técnica de MLST aporta la ventaja de la globalización, ya que los datos obtenidos se almacenan fácilmente en bases de datos accesibles vía Internet10. Este hecho permite crear una nomenclatura común de consenso sobre los diferentes genotipos y clones detectados en diferentes países.
En un trabajo previo realizado por nuestro grupo, el desarrollo de un nuevo esquema de MLST para E. faecalis ha permitido obtener los primeros datos acerca de la epidemiología global y estructura poblacional de esta bacteria7. En dicho estudio, se detectó la presencia de dos importantes complejos clonales (CC) denominados CC2 y CC9 que incluían cepas de origen hospitalario con resistencia a vancomicina y causantes de brotes hospitalarios. Posteriormente, CC2 y CC9 han sido definidos como complejos clonales de alto riesgo11 por su especial adaptación al ambiente hospitalario y su dispersión en diferentes países y continentes.
El objetivo del presente trabajo ha sido aplicar la técnica de MLST en cepas de E. faecalis sensibles y resistentes a la vancomicina aisladas en España para conocer su estructura poblacional y definir la posible presencia de ambos CC de alto riesgo en nuestro país.
Material y métodos
Cepas estudiadas
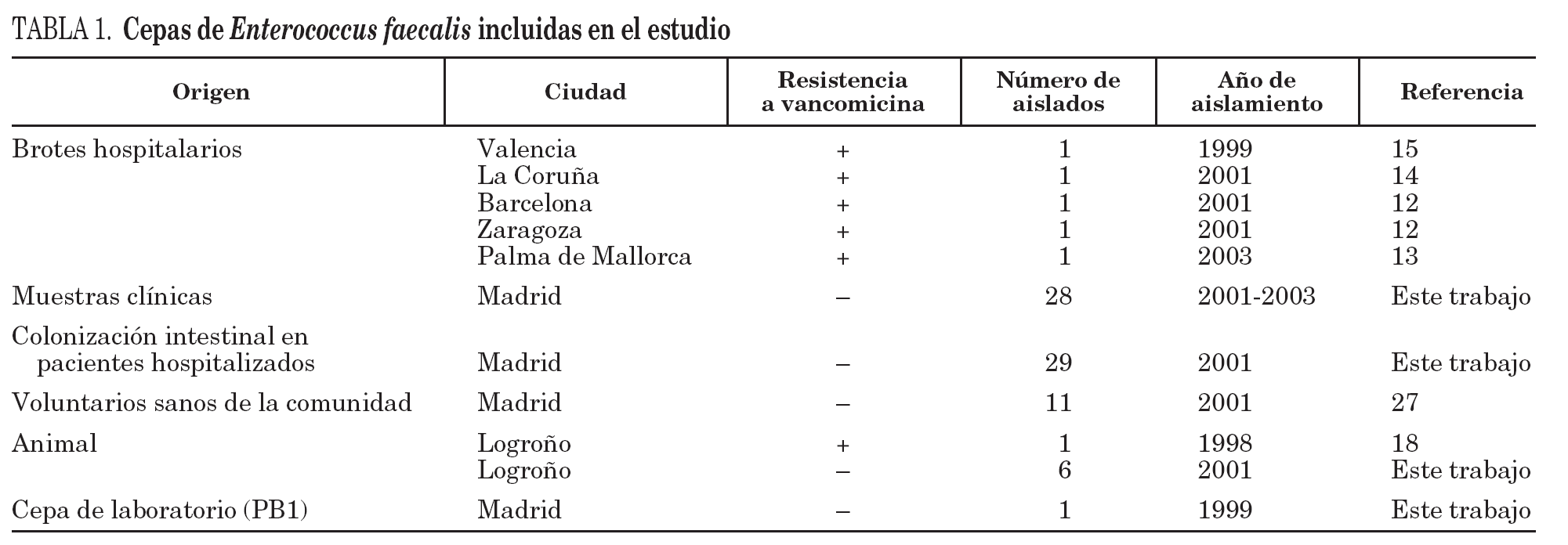
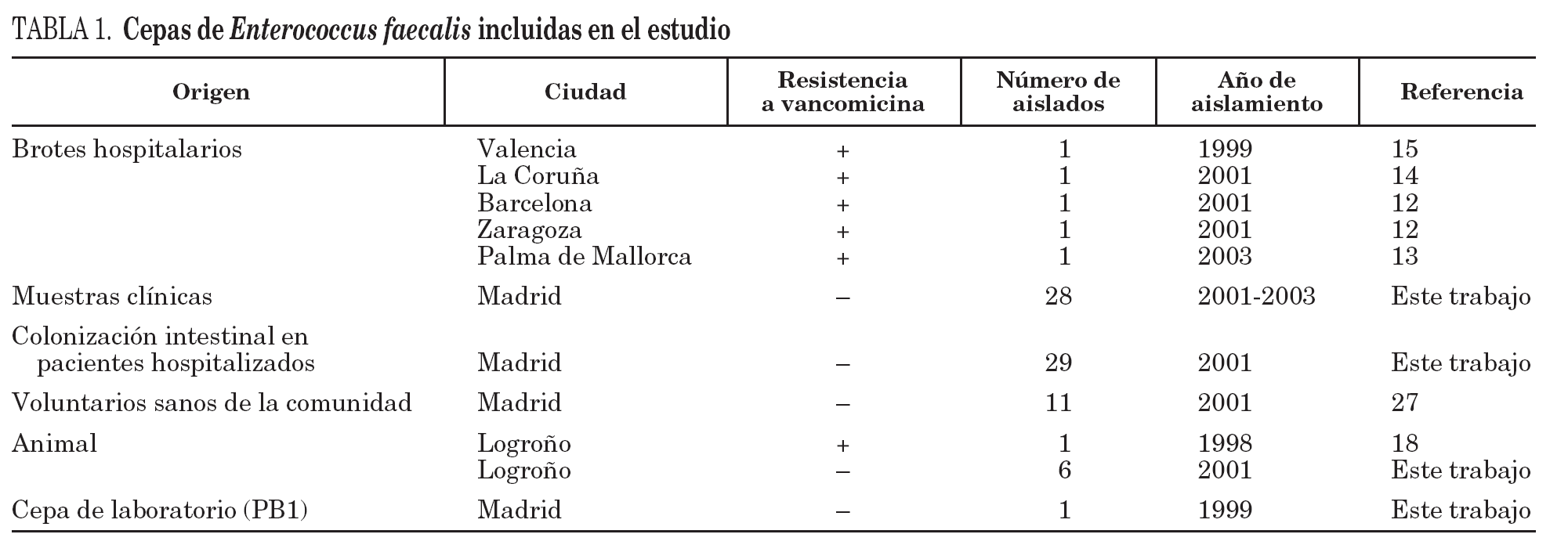
Se han incluido 81 cepas de E. faecalis procedentes de siete regiones españolas con diferentes orígenes y recogidas en distintos años (tabla 1). En esta colección también se han incluido cepas de E. faecalis resistentes a la vancomicina causantes de los brotes hospitalarios descritos hasta la fecha en España12-15 debido a su gran relevancia clínica y epidemiológica. Para la identificación de la especie se emplearon los cebadores específicos para el gen que codifica la D-alanina: D-alanina ligasa (ddl)16.
Multilocus sequence typing
Se amplificó mediante reacción en cadena de la polimerasa (PCR) fragmentos internos de 7 genes metabólicos muy conservados (gdh, gyd, pstS, gki, aroE, xpt y yiqL), siguiendo el esquema previamente propuesto7 (http://efaecalis.mlst.net). Todos los productos de amplificación fueron finalmente secuenciados en ambas direcciones para su posterior análisis.
Análisis de los datos
A cada nueva secuencia o alelo identificado para cada uno de los 7 genes que componen el esquema se les asignó un número en función del orden de aparición y en concordancia con la base de datos de MLST para E. faecalis (http://efaecalis.mlst.net). Posteriormente, a las combinaciones de los siete alelos se les asignó un perfil alélico o tipo de secuencia (sequence type [ST]) utilizando los genes en el siguiente orden: gdh, gyd, pstS, gki, aroE, xpt, yqiL (http://www.mlst.net).
La relación entre las diferentes ST se muestra en un dendrograma construido con el método UPGMA empleando el programa informático START (http://www.mlst.net). Las ST se agruparon en CC mediante el programa informático eBRUST17 (http://www.mlst.net). Los CC se definen como grupos de ST relacionadas entre sí que comparten al menos 5 de los 7 alelos con otras ST del mismo grupo. Además, los CC llevan implícito un concepto evolucionista ya que estas ST se han diversificado recientemente a partir de un antecesor común. El programa informático eBRUST permite determinar la ST que con mayor probabilidad representa el genotipo fundador de un CC. Se considera como posible ST fundadora de un CC a aquella que presenta un mayor número de ST dentro del mismo grupo que se diferencian entre sí en un solo alelo (single locus variants [SLV]). Por último, las ST que no pueden asignarse a ningún CC se denominan singletons.
Evaluación de la recombinación
El índice de asociación (Ia)18 estima el equilibrio de ligamiento entre los diferentes alelos de los 7 loci. El Ia se define como: Ia = VO/VE 1, en el que VO es la varianza observada de K y VE es la varianza esperada de K, y K es el número de locus en el que se diferencian dos individuos. Cuando los alelos están en equilibrio de ligamiento el Ia tiene un valor próximo a cero y, por tanto, es una población en la que la recombinación es frecuente. Por el contrario, una población clonal se caracteriza por presentar un Ia que se desvía significativamente de cero. Para calcular el Ia se utilizó el programa informático START (http://www.mlst.net).
Resultados
Relación filogenética entre los distintos aislados de E. faecalis
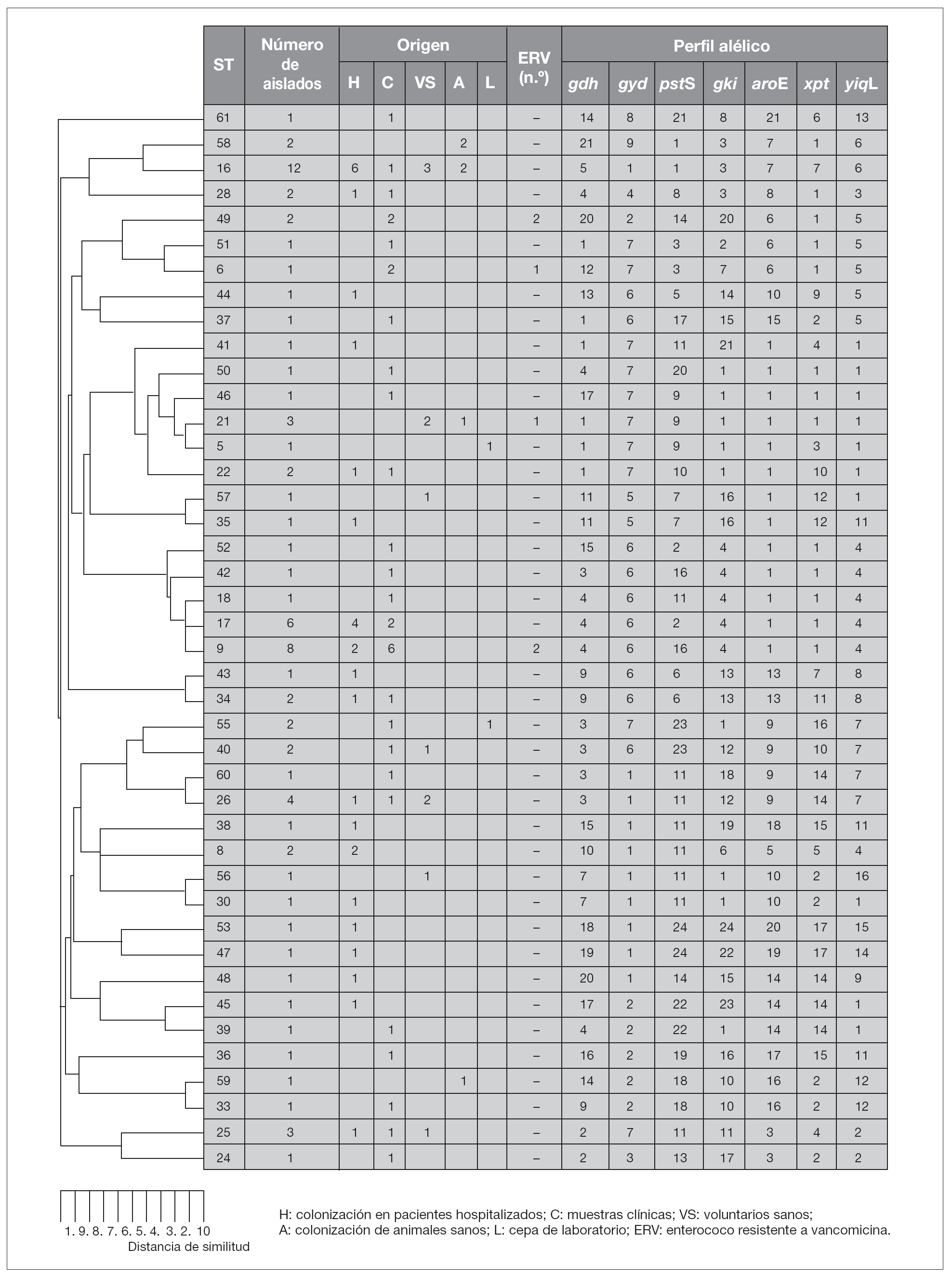
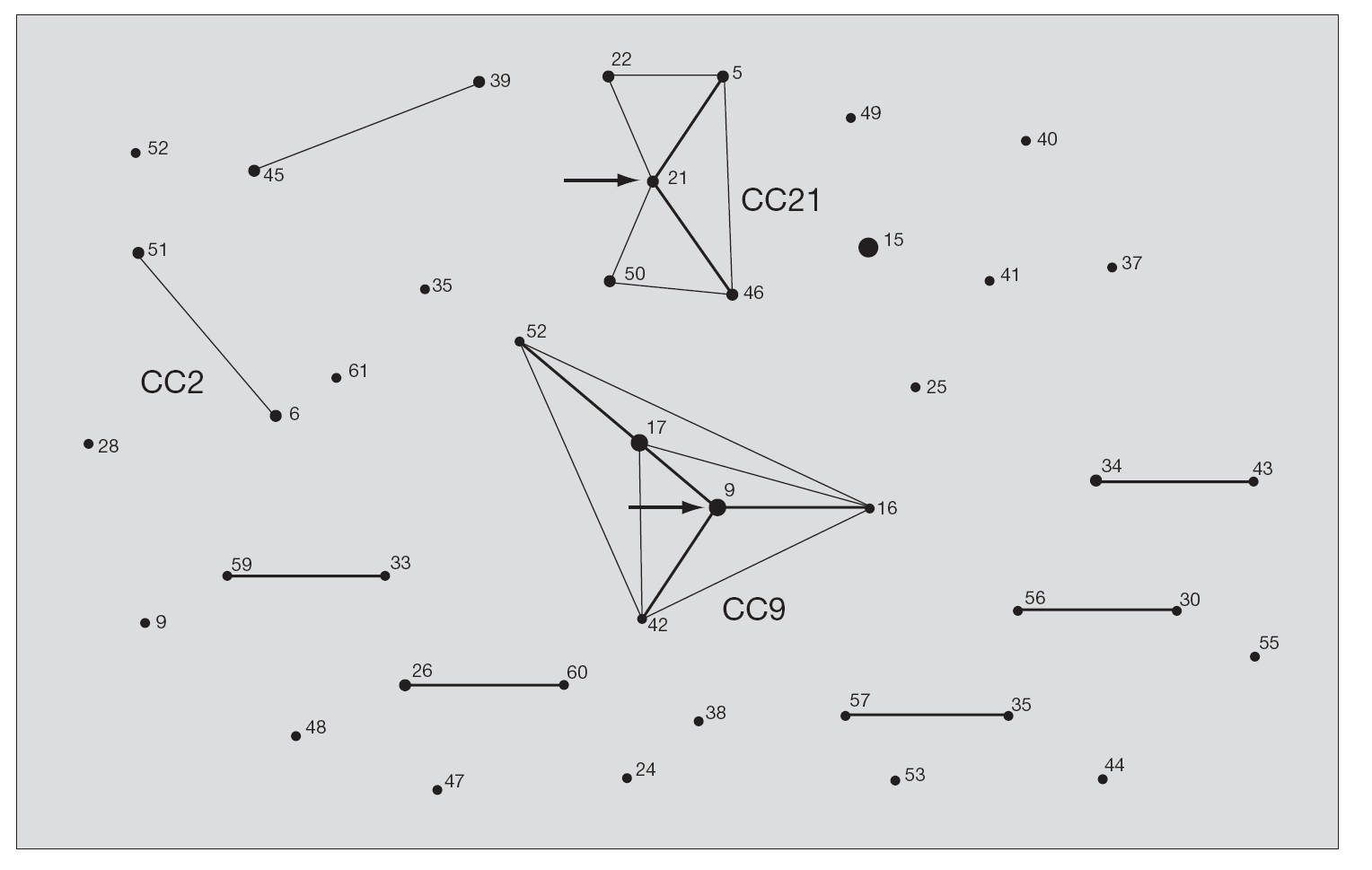
Tras la caracterización de las 81 cepas de E. faecalis empleando MLST, se definieron un total de 42 ST. Las ST que agruparon un mayor número de aislados fueron: ST16 (12 aislados), ST9 (8 aislados) y ST17 (6 aislados) y ST26 (4 aislados), mientras que, por el contrario, 28 de las 42 ST se correspondieron con un único aislado. Las relaciones filogenéticas entre las 42 ST se han representado en un dendrograma mediante el método UPGMA (fig. 1). Empleando el programa eBRUST, las 42 ST se agruparon en 2 CC mayores, 7 CC menores y 18 singletons. Los CC mayores se designaron como CC9 (17 aislados) y CC21 (8 aislados) ya que en ambos complejos fue posible asignar a la ST9 y ST21 como fundadoras del grupo. De los 18 singletons, 8 agruparon más de un aislado, y fue la ST16 la que concentró a un mayor número de ellos (n = 12). En la representación gráfica de eBRUST se muestran todas las ST agrupadas en sus correspondientes CC y las relaciones que se establecen (SLV o double locus variants [DLV]) entre las mismas (fig. 2).
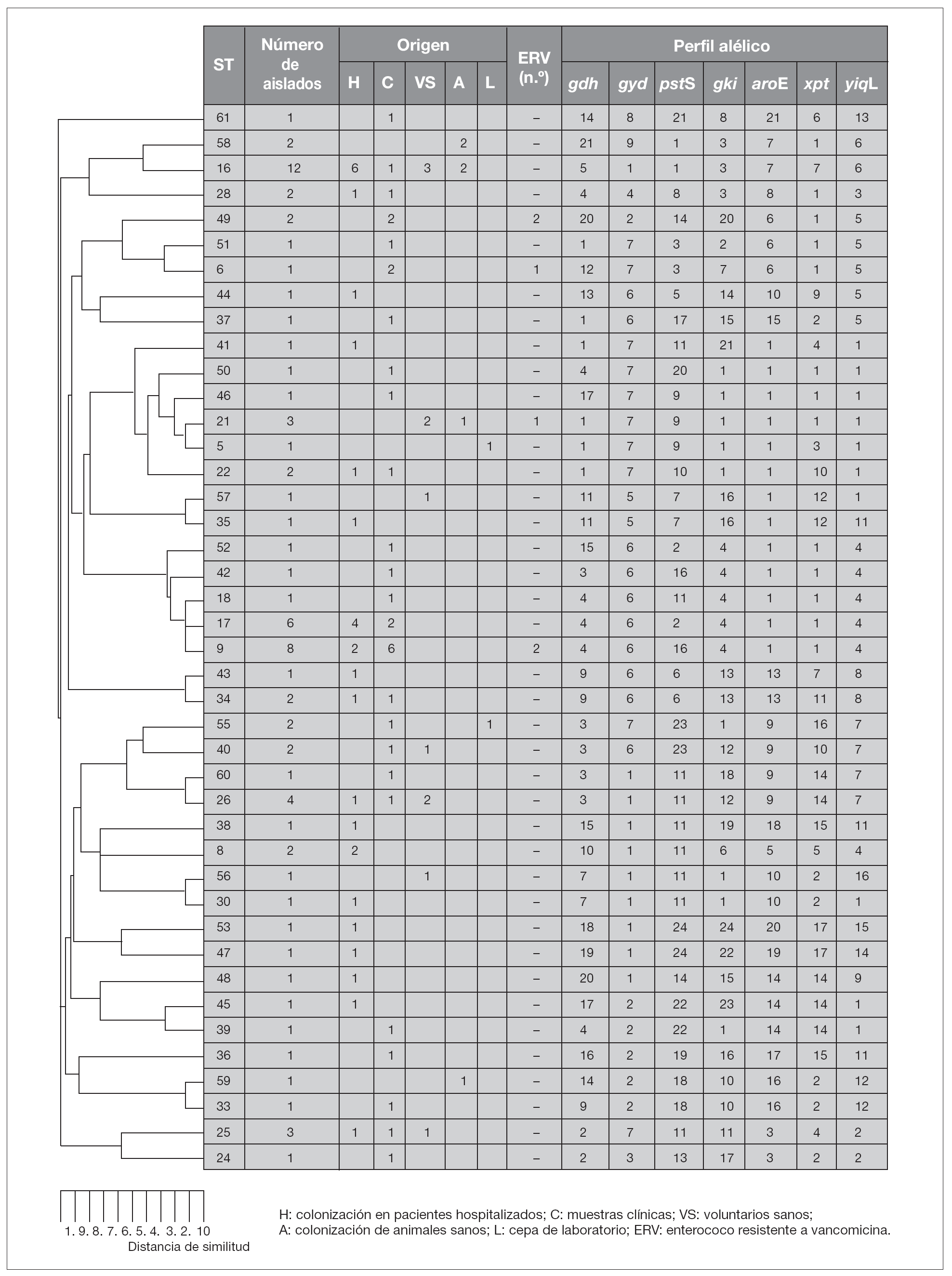
Figura 1. Relación filogenética entre los 42 ST (tipos de secuencia) representados en un dendrograma mediante el método UPGMA.
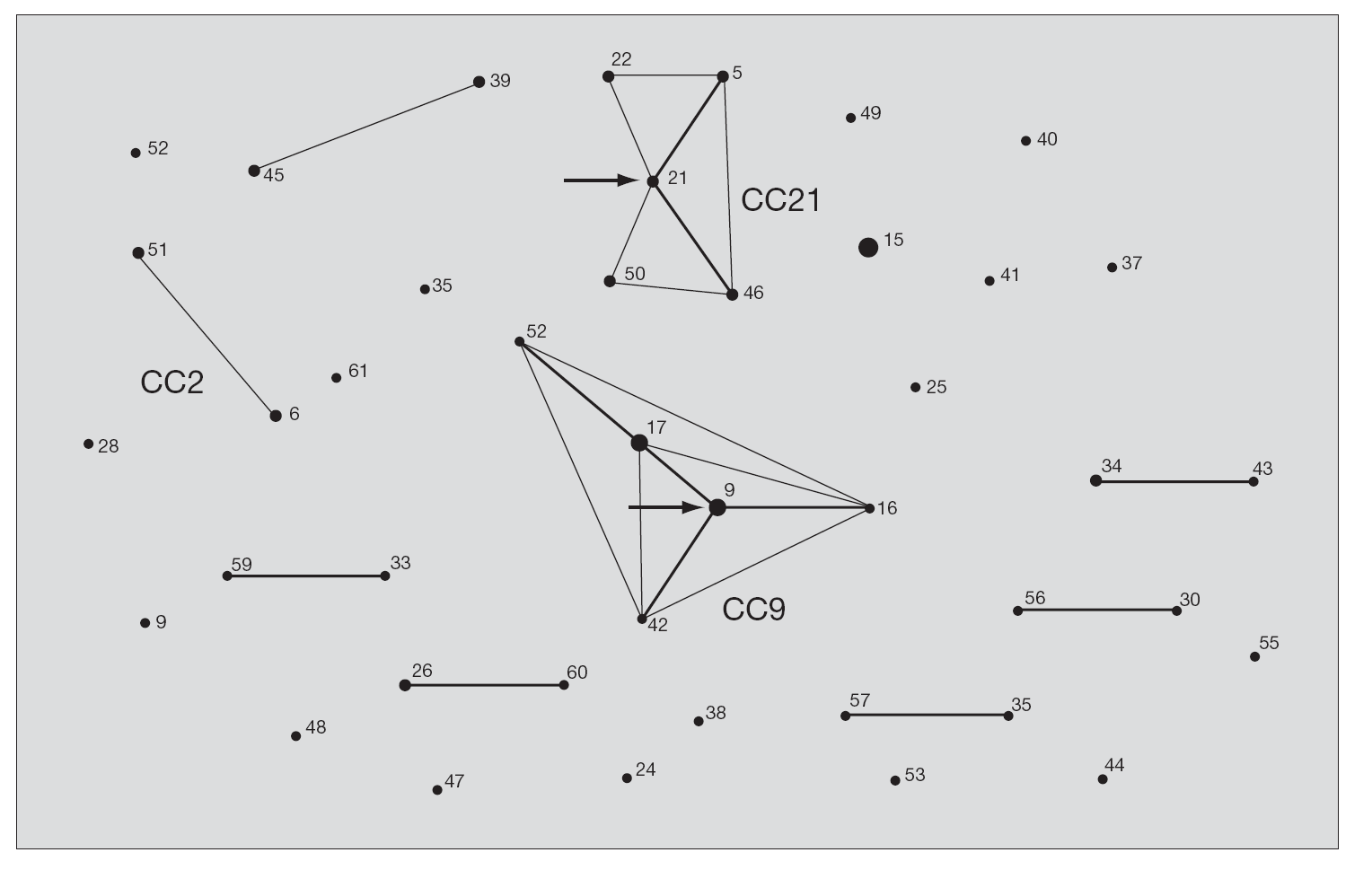
Figura 2. Representación de los complejos clonales mediante eBURST. Los números corresponden a cada ST. El área de cada círculo se corresponde con el número de cepas que se agrupan en cada ST. Las líneas grises finas conectan ST que se diferencian en un alelo (SLV) y las líneas grises gruesas conectan ST que se diferencian en dos alelos (DLV). Las flechas señalan las posibles ST antecesoras de CC9 y CC21. Los complejos clonales mayoritarios en la población de E. faecalis estudiada en España (CC9 y CC21), así como los de alto riesgo hospitalario (CC2 y CC9), aparecen señalados en la figura.
Epidemiología de los complejos clonales
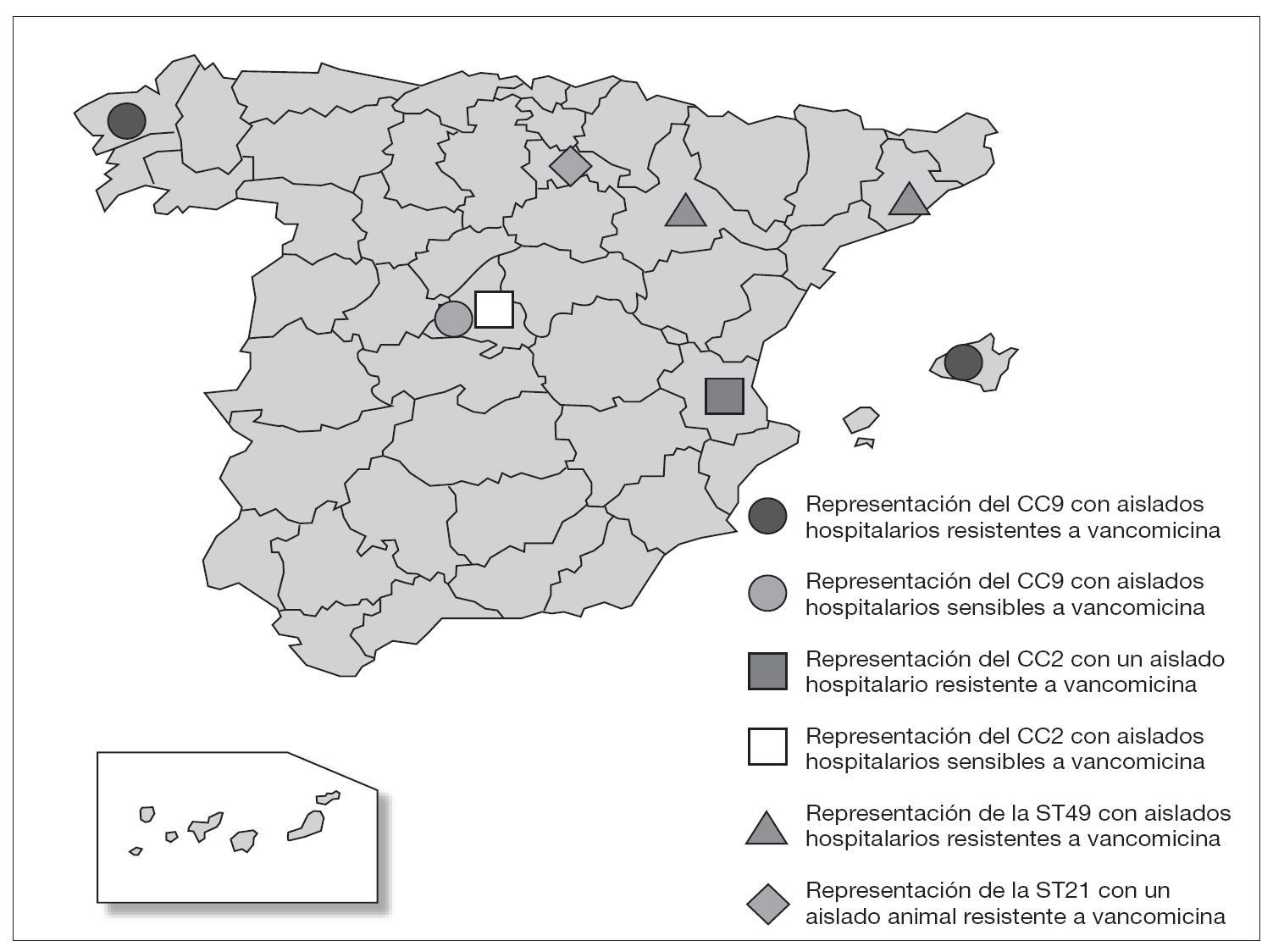
Los aislados de E. faecalis caracterizados con este esquema de MLST se agruparon en CC independientemente de su origen epidemiológico y geográfico, y fue posible encontrar en un mismo complejo cepas de origen animal y hospitalario (fig. 1). Un ejemplo de esta situación es el singleton clon ST16 (12 aislados), en el que se concentran cepas procedentes de distintas regiones (Madrid [n = 10] y La Rioja [n = 2]) y diversos orígenes epidemiológicos (muestras clínicas [1], colonización intestinal de pacientes hospitalizados [6] y de voluntarios sanos de la comunidad [3], así como de animales [3]) (fig. 1). A pesar de que no parece existir una aparente relación específica entre ciertas ST y CC con determinados orígenes epidemiológicos, en un estudio previo se definieron dos complejos, denominados CC2 y CC9, especialmente adaptados al ambiente hospitalario7. En este trabajo, en el que se han analizado aislados procedentes de diversas regiones españolas, se ha detectado la presencia de CC9 que agrupó cepas de origen exclusivamente hospitalario (17 de 57, 28% de cepas aisladas a partir de muestras clínicas y colonización de pacientes hospitalizados), entre las que se incluían aislados de E. faecalis resistentes a vancomicina causantes de brotes hospitalarios en Palma de Mallorca13 y La Coruña14 (figs. 1 y 3). Por otra parte, la cepa resistente a glicopéptidos procedente de un brote hospitalario en Valencia15 se agrupó en la ST6 que pertenece a CC2, y los aislados procedentes de Zaragoza12 y Barcelona12 en la ST49 (figs. 1 y 3). La cepa de origen animal resistente a vancomicina B343 19 se agrupó en el CC21 junto con otras cepas de origen animal y cepas procedentes de colonización intestinal de voluntarios sanos de la comunidad y pacientes hospitalizados, así como de origen clínico (fig. 1).
Evaluación de los procesos de recombinación en E. faecalis
Para conocer si la diversidad genética en E. faecalis se debe principalmente a procesos de mutación o bien de recombinación, se calculó el Ia para cuantificar el equilibrio de ligamiento entre los distintos alelos. En las poblaciones de tipo clonal en las que la variabilidad genética es debida principalmente a procesos de mutación, se muestran asociaciones entre los alelos de los diversos loci (desequilibrio de ligamiento). Por el contrario, en las poblaciones donde predominan los fenómenos de recombinación se detectan bajos grados de asociación entre los alelos de los diferentes loci (equilibrio de ligamiento). Al calcular el Ia para las 81 cepas y las 42 ST se obtuvo un valor de 2,7 y 1,34, respectivamente, lo que indica que en la población existe un desequilibrio de ligamiento. Cuando se volvió a calcular Ia incluyendo únicamente las ST no relacionadas entre sí genéticamente (ST que se diferencian en 3 o 4 loci y pertenecen a distintos CC), se obtuvo un valor de Ia próximo a cero (Ia = 0,42). La caída del valor del Ia cuando se calcula sólo para los genotipos no relacionados entre sí, frente al valor obtenido cuando se calcula para toda la población o ST, es propio de una estructura poblacional de tipo epidémico.
Discusión
Recientemente se ha descrito un nuevo esquema de MLST para E. faecalis basado en la amplificación y posterior secuenciación de fragmentos internos de 7 genes que codifican enzimas implicadas en rutas metabólicas, también conocidos como genes housekeeping7. La caracterización de 81 cepas de E. faecalis aisladas en España empleando este esquema de MLST ha permitido definir la presencia y dispersión de los complejos CC2 y CC9 en nuestro país.
Estudios realizados en una especie estrechamente relacionada como es E. faecium pusieron de manifiesto que en su estructura poblacional existía un fenómeno descrito como especificidad de origen, es decir, cepas aisladas a partir de determinados orígenes epidemiológicos se agrupaban en líneas genéticas concretas6. Por el contrario, este fenómeno no ha sido detectado en E. faecalis, de tal manera que cepas de diversos orígenes pueden agruparse en una misma ST y/o CC, como sucede en el caso de la ST16 y el CC21, que están presentes tanto en el medio hospitalario como en la comunidad colonizando a humanos y animales.
A pesar de la falta de especificidad de origen en E. faecalis, los primeros datos sobre su estructura poblacional pusieron de manifiesto la presencia de dos CC denominados CC2 y CC9 especialmente adaptados al medio hospitalario y muy diseminados por diversos países y continentes7,20. Estos complejos han sido descritos como CC de alto riesgo20, ya que en ellos se agrupan aislados de gran importancia clínica y epidemiológica, como es el caso de la cepa secuenciada de E. faecalis V583, de todas las cepas descritas hasta el momento productoras de betalactamasa7, así como de la mayor parte de las cepas resistentes a glicopéptidos causantes de brotes hospitalarios en diversos países y continentes (Ruiz-Garbajosa et al, datos no publicados). En España, entre los aislados de origen hospitalario analizados se han detectado ambos complejos, y existe una mayor representación de CC9 frente a CC2. Los aislados resistentes a vancomicina causantes de los brotes hospitalarios en Palma de Mallorca13 y La Coruña14 se agrupan en CC9, mientras que el aislado procedente del brote en Valencia15 se agrupa en CC2 (fig. 3). Por otra parte, en la ST49 se agruparon los aislados resistentes a glicopéptidos pertenecientes a un clon diseminado en Zaragoza y Barcelona12. La ST49 comparte 3 alelos con las ST agrupadas en CC2, lo que podría indicar una posible relación con este CC (fig. 1).
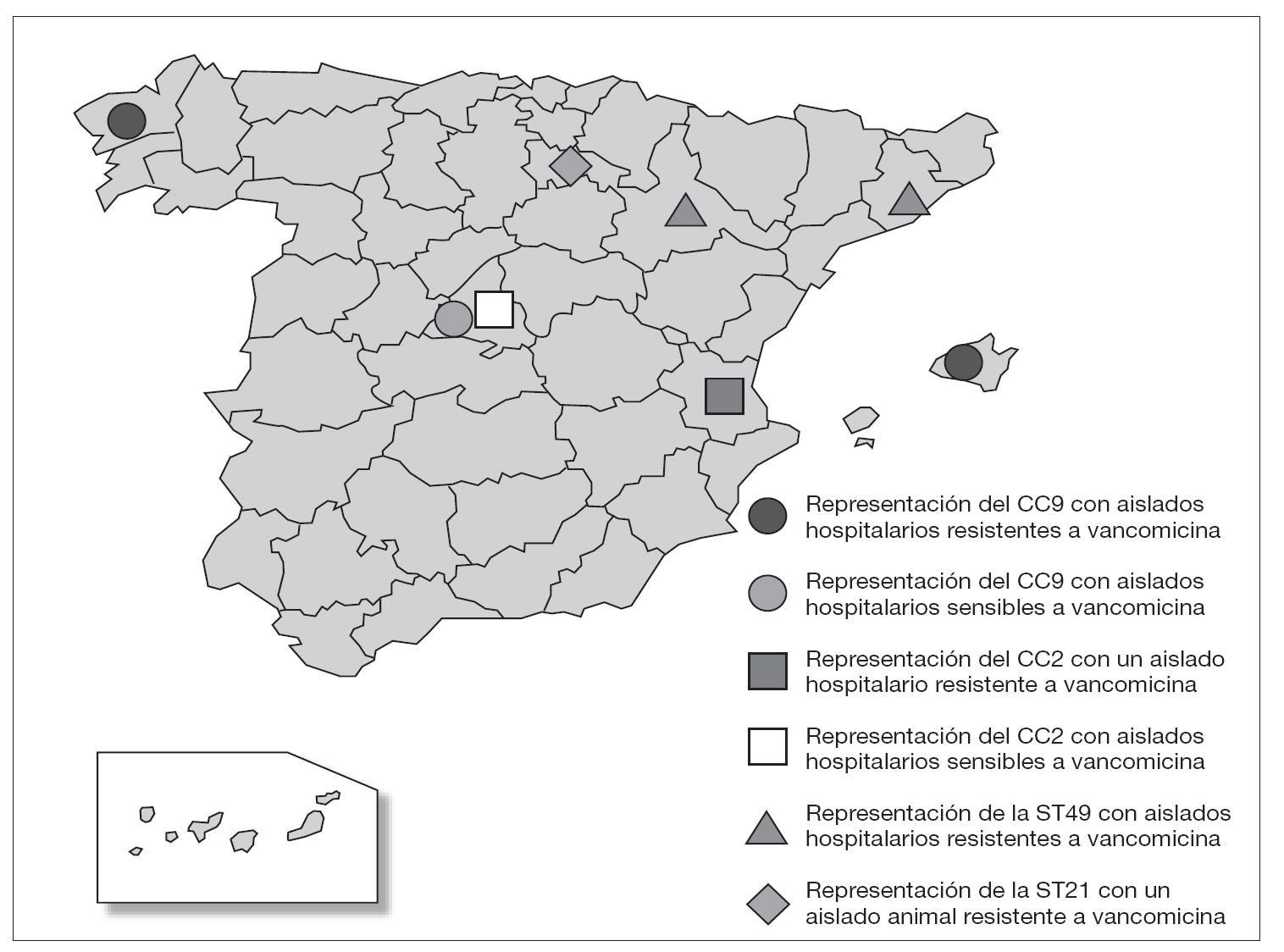
Figura 3. Representación de la localización en España de los complejos clonales de alto riesgo hospitalario CC2 y CC9 y de las ST que no pertenecen a estos CC y que representan cepas resistentes a la vancomicina.
Es importante destacar la presencia de CC9 y CC2 en Madrid, que agrupa aislados hospitalarios sensibles a vancomicina (fig. 3). Este hecho cobra gran importancia, ya que en numerosos estudios se ha postulado que ciertos clones endémicos21-25 pueden servir de sustrato para la posterior adquisición de la resistencia a vancomicina. Por ejemplo, en E. faecium se ha descrito el CC17 como complejo clonal de alto riesgo en el que se agrupan los aislados resistentes a vancomicina causantes de brotes hospitalarios. Además, los aislados sensibles que se agrupan en él se van a caracterizar por presentar resistencia a ampicilina y contener una isla de patogenicidad con el gen esp6; ambas características se comportan como mecanismos adaptativos. En el caso de E. faecalis CC2 y CC9, al igual que ocurre con CC17 en E. faecium, son complejos que han ido adquiriendo de forma secuencial mecanismos adaptativos como la resistencia a antibióticos y factores de virulencia. Todo ello favorece su persistencia en el medio y, a su vez, facilitan la adquisición de nuevos mecanismos. Este proceso se conoce como capitalismo genético26 y su resultado es una población seleccionada muy adaptada al ambiente hospitalario. Por otra parte, la adquisición de la resistencia a vancomicina es un paso más en este proceso de adaptación y puede tratarse de un fenómeno local relacionado con el consumo de antibióticos en cada hospital o área de estudio. De esta forma, los complejos clonales, como ha sucedido en España, pueden evolucionar de manera independiente en cada ambiente (hospital, comunidad, etc.), dependiendo de la disponibilidad genética en los mismos y de la presión selectiva ejercida por la utilización de antibióticos26.
Los procesos de recombinación han sido evaluados mediante el cálculo del Ia. La obtención de un valor del Ia próximo a cero cuando se calcula para los genotipos de la población no relacionados entre sí, frente a un valor más elevado cuando en el estudio se incluyen todas las ST o los 81 aislados, es característico de una población de tipo epidémico. En estas poblaciones la diversidad genética es generada por procesos de recombinación, pero en determinadas circunstancias emergen ciertos complejos clonales (como en este caso CC9) que, por una ventaja selectiva, se comienzan a expandir haciendo que estos genotipos predominen sobre el resto y creando una falsa apariencia de clonalidad en la población. El gran potencial de recombinación en E. faecalis puede favorecer y potenciar el capitalismo genético de estos complejos de alto riesgo. Además, aumenta la posibilidad de transmisión a otros CC, facilitando la dispersión de las resistencias. Por este motivo la resistencia a vancomicina puede también encontrarse en líneas genéticas no relacionadas entre sí, como sucede con la cepa de origen animal B43418, que se agrupa en el CC21.
La aplicación del esquema de MLST para E. faecalis nos ha permitido conocer los primeros datos acerca de su estructura poblacional en España y su relación en el ámbito de una epidemiología global y a largo plazo. Se ha detectado la presencia en nuestra geografía de dos CC, como son CC2 y CC9, dispersos también en otros países7,11,20 y de alto riesgo hospitalario, que localmente pueden evolucionar de forma diferente dependiendo de la carga genética del entorno al que se encuentren sometidos. En el futuro, las estrategias para el control de la infección en los hospitales deberían ir encaminadas a la detección precoz de los CC de alto riesgo epidemiológico. Esta actitud puede ayudar a predecir tendencias futuras en la aparición de resistencias, como el caso de la resistencia a vancomicina.
Agradecimientos
Este trabajo ha sido financiado parcialmente con los proyectos LSHM-CT-2007-037410 de la Unión Europea y PI061141 del Instituto de Salud Carlos III del Ministerio de Sanidad y Consumo de España.
Patricia Ruiz-Garbajosa está contratada (CM04/00013) a través del programa post-MIR (Formación en Investigación para Profesionales con Formación Sanitaria Especializada) del Instituto de Salud Carlos III del Ministerio de Sanidad y Consumo de España. Parte de este trabajo fue realizado en el laboratorio del Dr. R. Willems, en Utrecht (Países Bajos), gracias a una beca para estancias en el extranjero de la SEIMC.
Agradecemos a los Drs. G. Bou, A. Oliver, C. Torres, B.E. Murray y M. Zervos el envío de parte de las cepas incluidas en este estudio.
Correspondencia: Dra. P. Ruiz-Garbajosa.
Servicio de Microbiología. Hospital Universitario Ramón y Cajal.
Ctra. de Colmenar Viejo, km 9,100. 28034 Madrid. España.
Correo electrónico: pruizg.hrc@salud.madrid.org
Manuscrito recibido el 20-11-2006; aceptado el 7-3-2007.

