



Describir una nueva mutación en el exón 5 del gen PSEN1 (E120G) asociada a enfermedad de Alzheimer (EA) de inicio precoz y patrón de herencia autosómico dominante.
Paciente y métodosEl probando era un varón en el que se inició la enfermedad a los 34 años con problemas de memoria y deterioro cognitivo progresivo. Su padre y una hermana presentaron deterioro cognitivo de inicio precoz. El estudio genético por single strand conformation polymorphism (SSCP) de una muestra sanguínea del probando no detectó anormalidades que indicaran mutaciones en PSEN1, PSEN2 y APP. En los estadios finales de la enfermedad, el paciente presentó crisis epilépticas y alteración de la marcha. El paciente falleció a los 44 años. Los exones 3-12 del gen PSEN1 fueron analizados por secuenciación directa utilizando ADN aislado del tejido cerebral congelado del probando.
ResultadosEl examen neuropatológico reveló abundantes placas seniles y ovillos neurofibrilares, junto con una angiopatía amiloidea severa. El nuevo estudio genético del gen PSEN1 realizado mediante secuenciación directa detectó la mutación E120G.
ConclusionesE120G es una nueva mutación en PSEN1, probable causa de EA de inicio precoz con patrón autosómico dominante. La ausencia de mutaciones en estudios genéticos de cribado (SSCP) no descarta que haya mutaciones. Se recomienda el estudio genético mediante secuenciación directa en los casos de EA de inicio precoz y patrón de herencia autosómico dominante.
To describe a novel mutation in exon 5 of the presenilin 1 gene (E120G) associated with early-onset autosomal dominant Alzheimer’s disease (AD).
Patient and methodsThe proband was a man who began with memory loss and progressive cognitive decline at the age of 34. His father and his sister suffered from early-onset cognitive decline. The genetic study performed on the blood sample using the single strand conformation polymorphism (SSCP) technique did not detect any abnormality suggestive of the presence of a mutation in PSEN1, PSEN2, and APP. In the last stage of the disease the patient had seizures and gait alteration. He died at the age of 44. Coding exons 3-12 of PSEN1 were studied by direct sequencing using isolated DNA from frozen brain tissue of the proband.
ResultsThe neuropathological examination showed the presence of frequent amyloid plaques and neurofibrillary tangles and severe amyloid angiopathy. The direct sequencing of the PSEN1 gene disclosed the presence of the E120G mutation.
ConclusionsE120G is a novel mutation in PSEN1 that probably causes early-onset autosomal dominant AD. Absence of genetic alterations in screening techniques (SSCP) does not rule out the presence of mutations. We recommend direct sequencing for the genetic study of patients with early-onset autosomal dominant AD.
Por el momento se han implicado tres genes como causantes de enfermedad de Alzheimer (EA) monogénica, caracterizada por un inicio precoz y un patrón hereditario autosómico dominante1. Estos tres genes son el de la proteína precursora de amiloide (APP), el de la presenilina 1 (PSEN1) y el de la presenilina 2 (PSEN2). Entre ellos, las mutaciones en el gen PSEN1 son las que con mayor frecuencia causan EA de inicio precoz con patrón autosómico dominante, pues hasta el momento se han descrito 177 mutaciones patogénicas en él2. El fenotipo clínico de los pacientes con mutaciones en PSEN1 es similar al observado en la EA esporádica con una edad de inicio más precoz (media, 44 años), si bien se han descrito casos con un fenotipo particular (EA y paraparesia espástica, variante frontal de la degeneración lobular frontotemporal, EA asociada a hematomas lobulares, etc.)3–5.
En este estudio describimos una nueva mutación en el exón 5 de PSEN1 en un paciente con EA de inicio precoz y patrón autosómico dominante.
PacienteVarón de 39 años derivado al programa de información y consejo genético para demencias monogénicas (PICOGEN) de la Unitat d’Alzheimer i altres trastorns cognitius (UATC) del Hospital Clínic de Barcelona para valorar estudio genético6. Diestro, escolaridad hasta los 14 años, enolismo de 20g/día, consumo frecuente de cannabis sin criterios de dependencia y fumador sin otros antecedentes médicos relevantes. Su esposa refería que el paciente presentó los primeros problemas cognitivos a los 34 años en forma de olvido de recados e información reciente y algún episodio de desorientación en el transporte público. Un estudio neuropsicológico posterior mostró un deterioro cognitivo difuso de predominio temporal y frontal. El diagnóstico clínico fue EA probable en estadio 4 de la Global Deterioration Scale (GDS), y se inició tratamiento farmacológico con donepezilo. En el momento de la visita en la UATC, el paciente era todavía autónomo para las actividades básicas de la vida diaria, si bien presentaba un evidente problema amnésico (se afeitaba tres veces al día) y no salía solo a la calle por miedo a perderse.
La historia familiar evidenció una demencia de inicio precoz con un patrón de herencia autosómico dominante. La edad de inicio en el padre y una hermana del afecto no es bien conocida, si bien fallecieron a los 54 y los 49 años, respectivamente, tras sufrir un cuadro de deterioro cognitivo de varios años de evolución.
La exploración inicial no mostraba ninguna focalidad neurológica. La tomografía computarizada (TC) craneal no objetivó hallazgos significativos y la TC por emisión monofotónica (SPECT) cerebral evidenció una discreta hipoperfusión temporoparietal izquierda.
Tras una sesión de consejo genético y la firma del consentimiento informado, se realizó la extracción sanguínea del probando. Se analizaron los exones 3–12 de PSEN1 y PSEN2 y los exones 16 y 17 de APP mediante single strand conformation polymorphism (SSCP), pero no se detectó ninguna anormalidad que indicara alteraciones genéticas en esos genes.
En el seguimiento el paciente presentó un deterioro cognitivo y motor progresivo, con aparición de trastornos conductuales (pérdida de iniciativa, síntomas depresivos, irritabilidad, hiperactividad y heteroagresividad ocasional). El paciente mostró elevada sensibilidad a los efectos secundarios de los fármacos antipsicóticos, con distonía axial. En fases más avanzadas aparecieron ecolalia y conductas de utilización. En ese momento la exploración neurológica mostró hiperreflexia e hipertonía/espasticidad de predominio derecho, reflejos primitivos de la línea media y de liberación frontal y trastorno de la marcha. En el periodo final el paciente presentó pérdida de la deambulación, disfagia severa, caquexia y crisis comiciales. El paciente falleció a los 44 años, tras 10 años de enfermedad. La familia del paciente realizó la donación del encéfalo al banco de tejidos neurológicos del Hospital Clínic-Universidad de Barcelona.
MétodosSe realizó el estudio neuropatológico del paciente mediante tinciones de hematoxilina-eosina y detección inmunohistoquímica de amiloide βA4, proteína tau fosforilada (AT-8), alfasinucleína y TDP43. La extracción de ADN de tejido cerebral congelado del probando se realizó mediante el Dneasy tissue kit (Qiagen). Los exones 3–12 del gen PSEN1 fueron analizados por secuenciación directa según descripciones previas7.
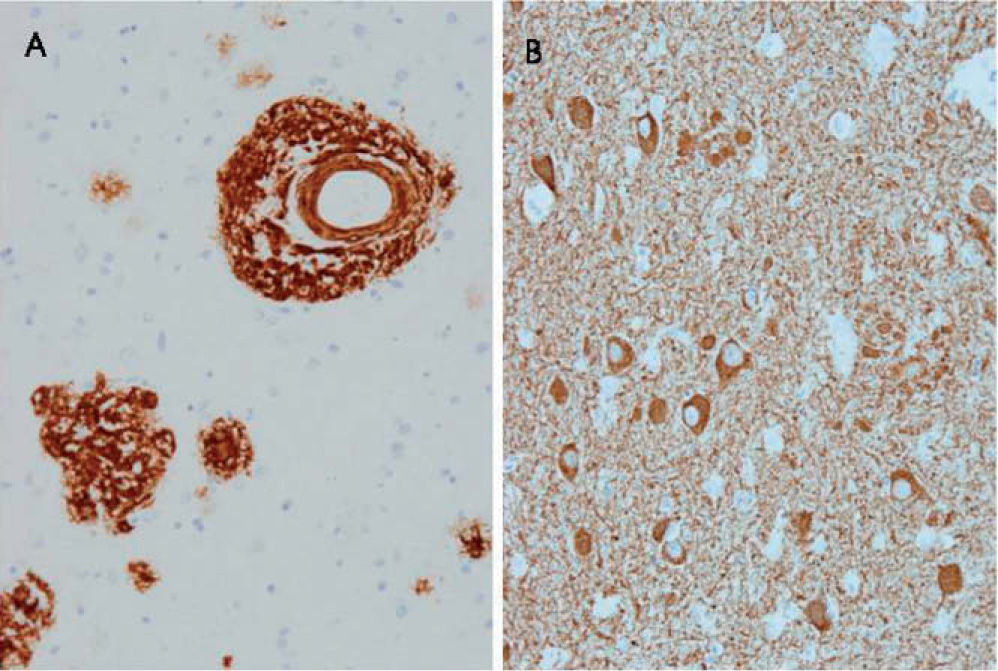
ResultadosEstudio neuropatológicoEncéfalo de 950g. En el estudio macroscópico se observaba marcada atrofia general. En el estudio microscópico se observaron numerosas placas seniles, difusas y maduras, con inmunorreactividad para βA4 de distribución difusa por todo el encéfalo, el tronco del encéfalo y el cerebelo (fig. 1). Junto a las placas seniles, presentaba inmunorreactividad de distribución generalizada para proteína tau fosforilada, en abundantes neuronas con degeneración neurofibrilar, placas y hebras neuríticas en el neuropilo (fig. 1). Ausencia de reactividad a alfasinucleína y TDP43. Angiopatía amiloide intensa en vasos leptomeníngeos e intracorticales, cerebrales y cerebelosos. El diagnóstico patológico final fue de EA estadio VI/VI de Braak y Braak de degeneración neurofibrilar y estadio C de depósitos de amiloide con intensa angiopatía amiloide asociada.

A: amiloide βA4; angiopatía amiloide cerebral y placas; los depósitos de amiloide del parénquima cerebral parecen difundirse de la pared de los vasos; también se observan placas de amiloide difusas y maduras. B: proteína tau foforilada; degeneración neurofibrilar en los citoplasmas de las neuronas y en abundantes procesos celulares o hebras en el neuropilo junto a placas neuríticas.
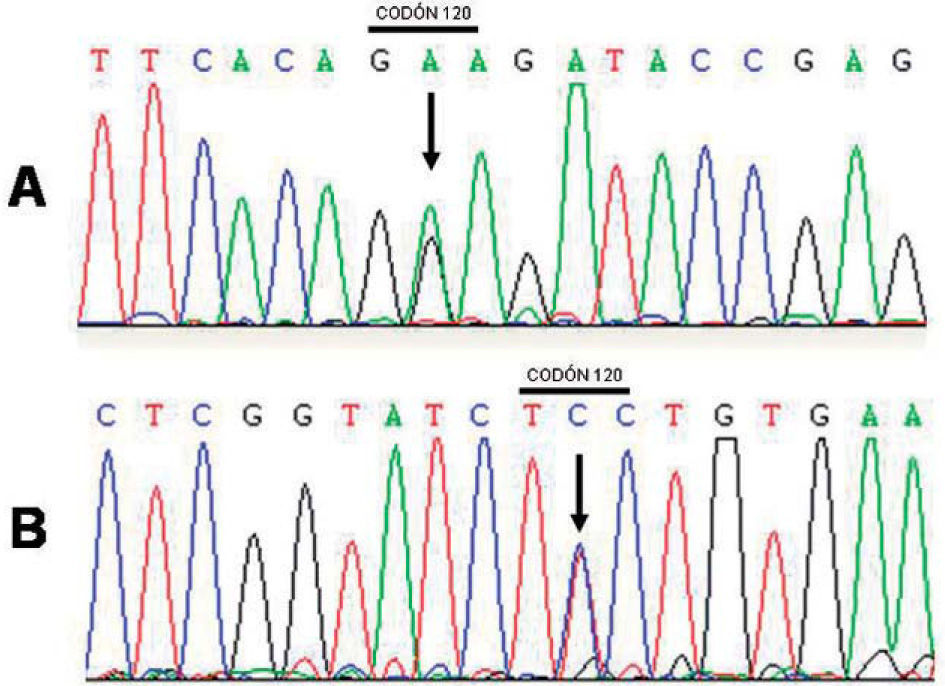
Se halló un cambio de nucleótido (A>G) en el exón 5 del gen PSEN1. Esta alteración genética conduce a un cambio de aminoácido en el codón 120 de glutamato (GAA) a glicina (GGA) que conduce a la mutación E120G (fig. 2). No se pudo realizar estudios de cosegregación de la mutación en esta familia, pues no existían otros familiares vivos afectos ni se disponía de tejido cerebral de los familiares afectos fallecidos.
Discusión. A: secuencia directa. B: secuencia inversa.")
La mutación E120G se sitúa en la primera región hidrófila que conecta los dominios transmembrana 1 y 2, en un codón altamente conservado en PSEN1 y PSEN28. Este hecho, junto con la presentación familiar de la enfermedad, la confirmación de EA en el estudio patológico y la descripción previa de otras mutaciones en el mismo codón (E120K, E120D)8,9, indica que tal mutación es la causante de la enfermedad en este paciente, y es improbable que se trate de un polimorfismo no patogénico infrecuente. Sin embargo, no se han podido realizar estudios de cosegregación ni estudios funcionales para confirmar su causalidad. En este sentido, según un artículo reciente de Guerreiro et al10 que evalúa el grado de patogenicidad atribuible a cambios genéticos detectados en los genes de PSEN1 y PSEN2, se habría de atribuir a esta mutación una patogenicidad en grado de probable.
La mutación E120G, al igual que la mayoría de las mutaciones en este gen descritas, conduce a una EA de inicio precoz con un patrón de herencia autosómico dominante. La edad de inicio precoz de este paciente es similar a la descrita en otra mutación del mismo codón (intervalo de la edad de inicio en mutación E120K, 32–39 años) y una década menos que en la otra mutación descrita en este codón (41-53 años)8,9. Las crisis epilépticas se han descrito en diversas mutaciones de PSEN1, entre ellas la mutación E120D. En la patología se detectan los hallazgos típicos de la EA junto con una importante angiopatía amiloide.
La aproximación técnica para la detección de mutaciones en pacientes candidatos se puede realizar de diferentes maneras, según las disponibilidades técnicas del laboratorio de diagnóstico. El estudio mediante secuenciación directa tiene una sensibilidad más elevada que el estudio por SSCP convencional (sensibilidad estimada, en torno al 80%, dependiendo del laboratorio concreto), si bien éste se utiliza con frecuencia como método de cribado inicial por su menor coste económico y de tiempo, y después se procede a secuenciar sólo los fragmentos que muestren un patrón anómalo11. Así pues, el clínico debe conocer la sensibilidad del estudio por SSCP al informar de los resultados al afecto y/o sus familiares. La existencia de falsos negativos en el estudio por SSCP debe hacer que el clínico se replantee la solicitud de un nuevo estudio genético por secuenciación directa en los casos con altas probabilidades de tener una mutación causante de EA en los que un estudio previo por SSCP haya resultado negativo5. El caso descrito enfatiza la importancia de realizar estudios de secuenciación en casos de EA de inicio precoz y patrón de herencia autosómico dominante, aun cuando el estudio por técnicas convencionales de cribado menos sensibles sea negativo. Sin embargo, se ha de subrayar que ninguna de estas técnicas permite evaluar la dosis génica, es decir, la presencia de deleciones o duplicaciones de todo el gen.
En resumen, describimos una nueva mutación en el gen de la PSEN1 (E120G) probable causa de EA de inicio precoz y con patrón de herencia autosómico dominante. Este caso enfatiza la importancia de realizar los estudios genéticos con técnicas de secuenciación directa en casos compatibles aun en casos negativos por técnicas menos sensibles.
FinanciaciónEste trabajo ha sido financiado por una beca de Pfizer-Eisai, proyecto FIS 080036 y Ajut a la Recerca Josep Font (Dr. Fortea).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores agradecen a los familiares del afecto la donación del tejido cerebral del paciente al Banco de Tejidos Neurológicos de la Universitat de Barcelona/Hospital Clínic.

