




El síndrome de Tolosa-Hunt (STH) es una entidad cuya etiología es idiopática y que forma parte del diagnóstico diferencial de la oftalmoplejía dolorosa. Se debe a una infiltración granulomatosa de la pared lateral del seno cavernoso y se caracteriza por uno o más episodios de dolor orbitario unilateral junto con parálisis de uno o más nervios oculomotores. Con la resonancia magnética (RM) se puede demostrar la infiltración granulomatosa del seno cavernoso. El tratamiento de elección son los corticoides y, una vez instaurado el tratamiento, el cuadro clínico se resuelve en menos de 72 h. Las recurrencias de STH ocurren hasta en la mitad de los pacientes incluso tras meses o años de un primer episodio y suelen ser ipsolaterales. Sin embargo, es posible la remisión espontánea del cuadro. En el presente trabajo presentamos un caso de síndrome de Tolosa-Hunt con remisión espontánea y recurrencia posterior.
Presentamos a una mujer de 31 años, natural de Perú, residente en España desde hace 5 años. Como antecedentes personales refería: parálisis facial periférica derecha idiopática en 2009, que se resolvió ad integrum, y migraña episódica sin aura de comienzo hace 2 años. Se encuentra en tratamiento con anticonceptivos orales. La paciente acude en octubre del 2011 al servicio de urgencias por diplopía y cefalea de una semana de evolución. La diplopía es binocular, más intensa en la mirada horizontal derecha y no se modifica con la distancia. La cefalea es hemicraneal derecha de característica pulsátil, se acompaña de fotofobia, es similar a sus crisis de migraña habituales y se incrementa progresivamente hasta alcanzar un 8/10 en la escala visual analógica. Refiere además parestesias en la región peribucal y mandibular derecha. Niega proptosis u otras alteraciones oculares.
La paciente presentó un episodio de similares características en agosto del 2011. El episodio duró una semana y se resolvió espontáneamente, sin secuelas. La exploración física y las constantes vitales son normales. En la exploración neuro-oftalmológica se evidencia una anisocoria, con una pupila derecha de 4mm e izquierda de 3mm, que no se modifica con la privación ni con la estimulación lumínica. Las pupilas son normorreactivas tanto en el reflejo lumínico como en el consensuado. Se observa además una ptosis derecha y una desconjugación de la mirada en posición primaria, con ojo derecho esotrópico e izquierdo hipertrópico (cover-uncover positivo, cover-cover negativo). Al examinar los movimientos oculares se pone de manifiesto una limitación para la abducción y supradextroversión del ojo derecho, con diplopía máxima en dichas posiciones. En días sucesivos, se añade limitación en la aducción, infradextroversión e infralevoversión del mismo ojo. El resto de la exploración neurológica, incluyendo fondo de ojo, agudeza visual y el resto de los pares craneales, es normal. En resumen, la exploración neurooftalmológica es congruente con una parálisis de los nervios craneales iii y vi derechos.
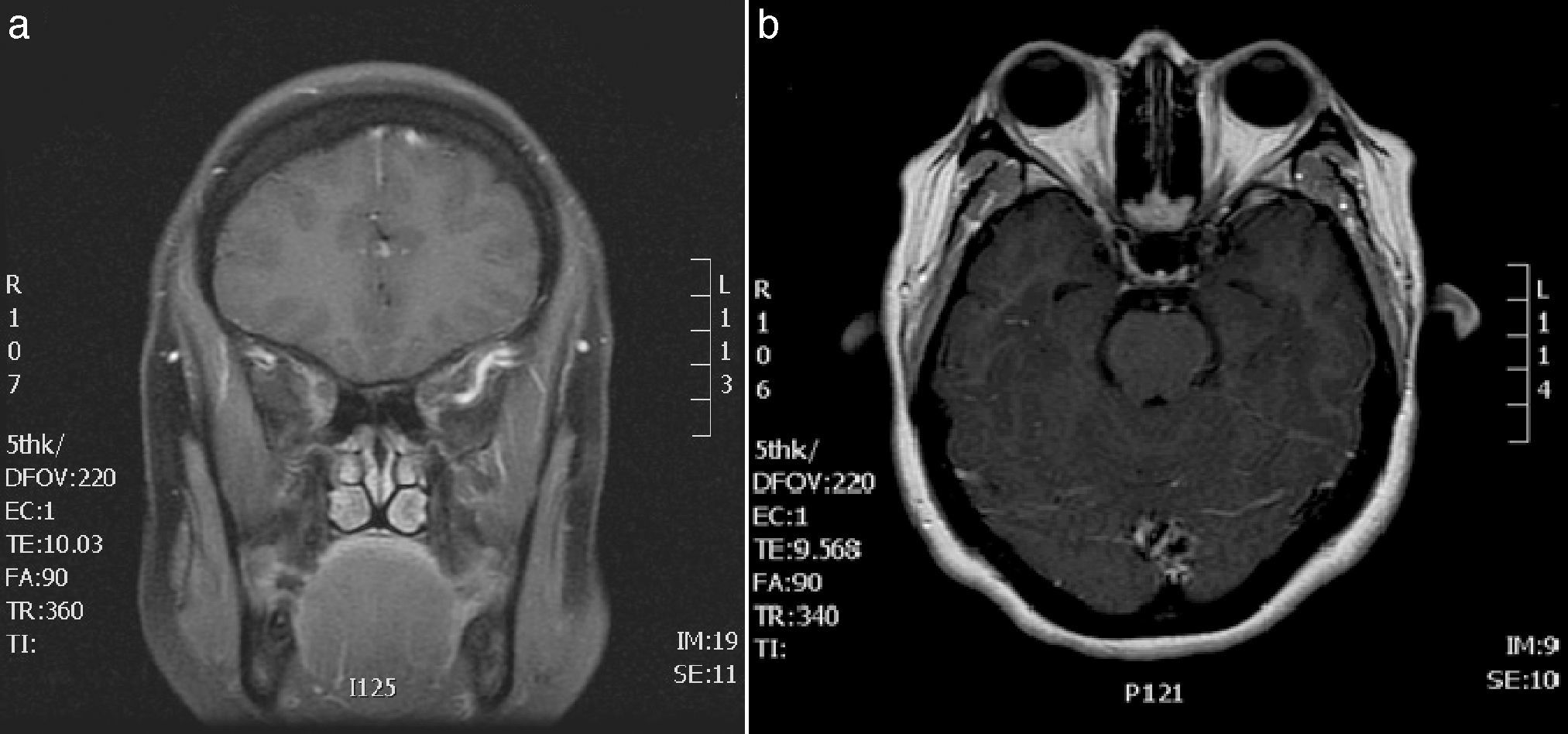
Con el cuadro clínico de la paciente se establece el diagnóstico sindrómico de oftalmoplejía dolorosa recurrente. Dentro del estudio se realiza hemograma y bioquímica (incluyendo velocidad de sedimentación globular [VSG] y enzima convertidora de angiotensinógeno [ECA]), objetivándose únicamente una hipertrigliceridemia. La serología de Borrelia y lúes, así como el estudio inmunológico (anticuerpos antinucleares, anticuerpos anticitoplasma de neutrófilos, antitiroideos), son negativos. Se realiza punción lumbar y análisis del líquido cefalorraquídeo incluyendo: citobioquímica, cultivo y Venereal Disease Research Laboratory, que no muestra alteraciones. En las imágenes obtenidas por RM, en las secuencias poscontraste, se evidencia realce asimétrico del ápex orbitario y de la pared lateral del seno cavernoso derecho (fig. 1). No existen asimetrías en el calibre ni en la intensidad de señal de los nervios ópticos.
 RM cerebral, secuencia T1 coronal con gadolinio. Se observa alteración de la señal en el seno cavernoso derecho. B) RM cerebral y de órbitas. Secuencia T1 axial con gadolinio. Se observa realce asimétrico del ápex orbitario y pared lateral del seno cavernoso derecho.")
Sobre la base a los hallazgos obtenidos en la neuroimagen más un cuadro clínico compatible y habiendo descartado otras etiologías, se considera el diagnostico de STH. Se inicia tratamiento con metilprednisolona 1g/24h por vía intravenosa durante 3 días y posteriormente pauta descendente de prednisona por vía oral. En las primeras 24 h tras la instauración de tratamiento, tanto la cefalea como la diplopía mejoran de forma progresiva hasta quedar la paciente asintomática.
El STH es un proceso inflamatorio del seno cavernoso de naturaleza idiopática. Es una patología infrecuente, que afecta por igual a ambos sexos y es más frecuente en la edad media de la vida. Algunos autores han descrito casos en niños1. Se caracteriza por la existencia de dolor orbitario unilateral, que puede irradiarse a la región retroorbitaria, frontal, temporal e incluso occipital. El dolor se asocia a diplopía por afectación de uno o más nervios oculomotores y también pueden afectarse la primera/segunda rama del trigémino, ipsolaterales2,3. La afectación de los pares craneales puede ocurrir simultáneamente a la cefalea o hasta 2 semanas más tarde. Se han descrito casos que asocian afectación de otros pares craneales como el ii, 3.ª rama del v, el vii o de la vía simpática pupilar ipsolateral4. La afectación bilateral es excepcional. En el año 2004, la international headache society hizo una revisión de los criterios diagnósticos iniciales de 19985.
La prueba de imagen de elección ante un paciente con oftalmoparesia dolorosa es la RM cerebral y de órbitas. En el STH se objetiva un engrosamiento del seno cavernoso isointenso en secuencias T1 /T2, y que realza con la administración de gadolinio. La ausencia de hallazgos en la RM no descarta la posibilidad de un STH, ya que se han descrito casos con clínica característica y neuroimagen normal6. El estudio con RM del seno cavernoso y el ápex orbitario tiene una alta sensibilidad para la detección y el seguimiento de la lesión inflamatoria del STH7.
El tratamiento de elección, incluido entre los criterios diagnósticos del STH5, son los corticoides. No hay datos definitivos sobre cuál es la dosis más adecuada, la frecuencia, la duración, la vía de administración del tratamiento, ni el tipo de corticoide. Las pautas utilizadas habitualmente son: bolos de metilprednisolona por vía intravenosa de 500-1.000mg/día durante 3-5 días o prednisona por vía oral a dosis de 1mg/kg día8. La duración del tratamiento es variable, desde semanas a meses, y se debe individualizar según la respuesta clínica del paciente. Los pacientes tratados con dosis más altas de esteroides (500-1.000mg/día durante varios días, después continuando con dosis de 1mg/kg al día varias semanas) podrían tener menor probabilidad de recurrencia9.
El STH es un diagnóstico de exclusión, siendo importante un buen diagnóstico diferencial10. Para ello, se debe realizar un estudio completo que incluya una analítica con VSG y ECA, radiografía de tórax, serología, inmunología, punción lumbar y estudio de neuroimagen. En el diagnóstico diferencial hay que considerar otras patologías que cursen con oftalmoparesia dolorosa y/o afecten al seno cavernoso, fisura orbitaria superior u órbita10,11. Igualmente, se deben tener en cuenta patologías que asocien cefalea y oftalmoparesia o diplopía fluctuante/episódica12 (tabla 1).
Entidades que cursan con diplopía fluctuante o episódica
| Miastenia gravis |
| Neuromiotonía |
| Mioquimia del oblicuo superior |
| Oftalmopatía distiroidea |
| Foria descompensada |
| Espasmo de convergencia |
| Espasmos cíclicos |
| Oftalmoplejía intermitente |
| Síndrome de Tolosa-Hunt |
Modificada de Porta-Etessam et al.13
En este caso clínico, dado el antecedente de migraña episódica de la paciente y un episodio previo autolimitado de similares características, nos plantemos como primera posibilidad diagnostica la migraña oftalmopléjica. Pero ante los hallazgos característicos en la RM y la resolución tanto del dolor como de la oftalmoparesia, en las primeras 12 h tras tratamiento con corticoides se llegó al diagnóstico de STH recurrente, tras un primer episodio con remisión espontánea.
Aunque existen remisiones espontáneas, son más frecuentes las recurrencias, que ocurren en el 40-50% de los pacientes y suelen ser ipsolaterales, algunas contralaterales13 o raramente bilaterales. Debido a la frecuencia de las recaídas en el STH, se recomienda el seguimiento posterior de estos pacientes. Las recaídas usualmente ocurren en los meses posteriores al primer episodio, aunque se han descrito casos más de una década después del primer episodio14. El seguimiento posterior, tanto clínico como radiológico, de estos pacientes es importante, ya que patologías como la sarcoidosis, las vasculitis o el linfoma pueden responder inicialmente al tratamiento con corticoides y es la evolución posterior la que nos lleva al diagnóstico correcto15.
La remisión espontánea referida por la paciente es una característica inusual del STH y son infrecuentes en la literatura científica de los últimos años. Los casos que están descritos son previos al uso de corticoides de forma habitual en el manejo del STH. Es probable que con los nuevos criterios diagnósticos, sumado a los avances en neuroimagen, se llegue a un diagnóstico de STH de forma más temprana, permitiendo así la instauración del tratamiento corticoideo de forma precoz, haciendo que las remisiones espontáneas sean poco frecuentes. No obstante, las remisiones espontáneas no son improbables. Por lo tanto, ante un paciente con un episodio de oftalmoparesia dolorosa que ha remitido espontáneamente y con recurrencia posterior debemos incluir dentro del diagnóstico diferencial el STH.

