



Nos gustaría exponer un caso inusual de deterioro neurológico repentino y coma. No hemos encontrado en la literatura ninguna descripción de debut clínico similar de esta enfermedad.
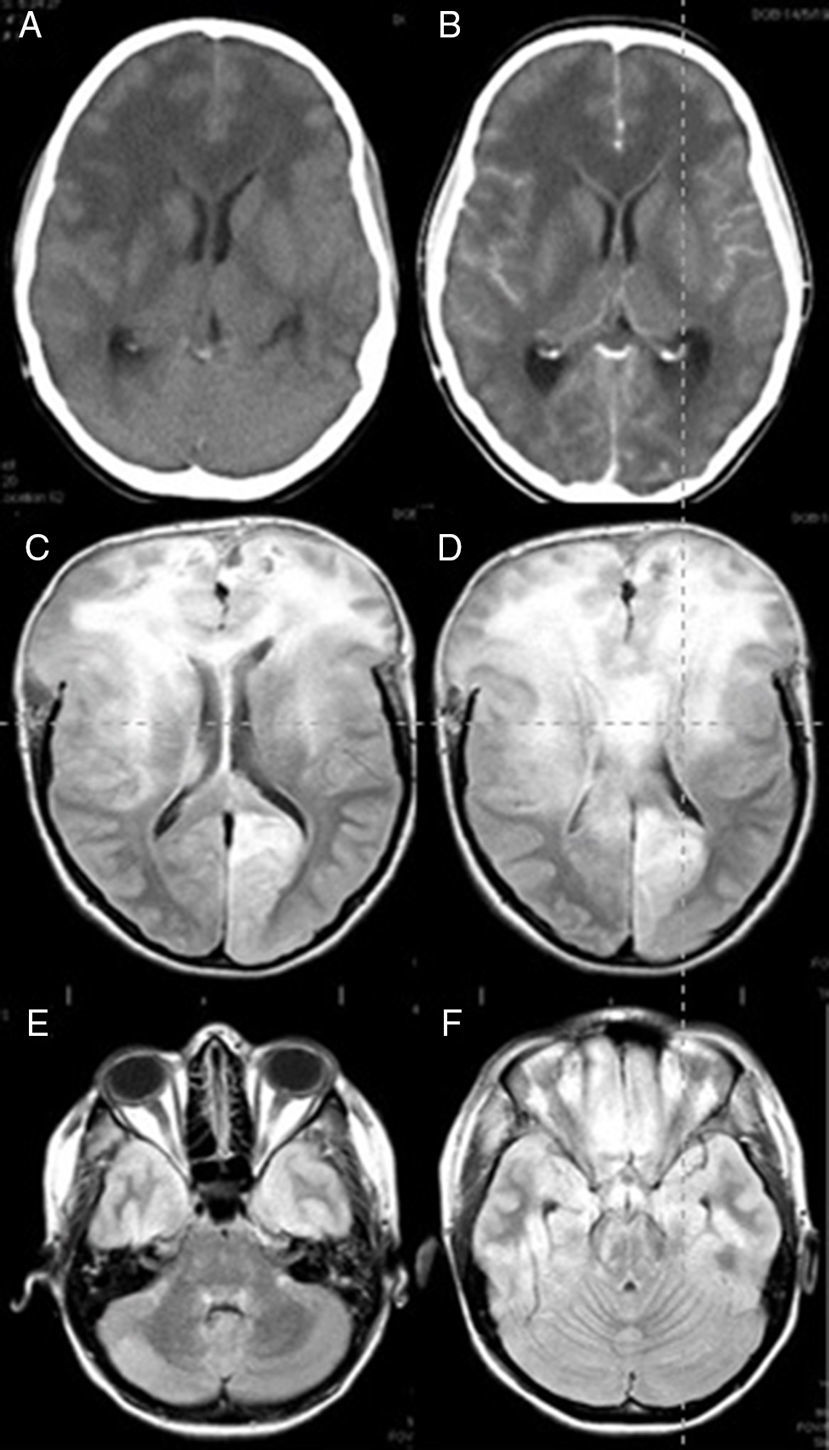
Nuestra paciente era una mujer de 25 años que había dado a luz 3 meses antes tras un embarazo controlado que había llegado a término sin complicaciones. Estaba en tratamiento con hierro por vía oral por una anemia posparto; no presentaba otros antecedentes médicos de interés. Se presentó en urgencias con un cuadro de cefalea, apatía y bradipsiquia de un mes de evolución. La cefalea había empeorado en las últimas 48 h y no respondía al tratamiento analgésico convencional. En las últimas horas había presentado náuseas y vómitos, pero no tenía fiebre. Mientras se encontraba en la sala de urgencias, sufrió una pérdida repentina de la consciencia y una crisis tónico-clónica, por lo que fue intubada. Se le realizó una tomografía computarizada de urgencia, que mostró signos de edema cerebral difuso y ocupación de las cisternas subaracnoideas. Se evidenció realce leptomeníngeo y ependimario (fig. 1A y B). Estos hallazgos eran compatibles con cerebritis acompañada de afectación meníngea y ventriculitis.
 y RM FLAIR (C-F). A) Presencia de hipodensidad difusa de la sustancia blanca predominantemente en los 2 lóbulos frontales y afectación del cuerpo calloso, que indican edema vasogénico. Se aprecia borramiento de los surcos de la convexidad. B) Imagen tomada tras administrar el contraste, que muestra ausencia de realce en las áreas mencionadas. C) Los signos de hipertensión intracraneal son especialmente evidentes en los lóbulos frontales y el cuerpo calloso. D) La herniación cerebral no desaparece a pesar de haber realizado una craniectomía bifrontal. E y F) Se aprecia afectación difusa también a nivel infratentorial.")
Imágenes de TC (A y B) y RM FLAIR (C-F). A) Presencia de hipodensidad difusa de la sustancia blanca predominantemente en los 2 lóbulos frontales y afectación del cuerpo calloso, que indican edema vasogénico. Se aprecia borramiento de los surcos de la convexidad. B) Imagen tomada tras administrar el contraste, que muestra ausencia de realce en las áreas mencionadas. C) Los signos de hipertensión intracraneal son especialmente evidentes en los lóbulos frontales y el cuerpo calloso. D) La herniación cerebral no desaparece a pesar de haber realizado una craniectomía bifrontal. E y F) Se aprecia afectación difusa también a nivel infratentorial.
La paciente fue ingresada en la Unidad de Cuidados Intensivos y a las 6 h presentó midriasis bilateral arreactiva de manera repentina. En este punto decidimos someterla a una craniectomía descompresiva bifrontal como tratamiento compasivo con el objetivo de descomprimir ambos lóbulos frontales y temporales. Asimismo realizamos una biopsia del lóbulo frontal derecho. Durante el procedimiento, observamos que el cerebro estaba congestivo y tenía una consistencia dura. Tras la operación, colocamos un sensor de presión intracraneal, que inicialmente registró una presión por debajo de los 15mmHg. Después de la craniectomía, la midriasis revirtió y la paciente recuperó los reflejos pupilares. El análisis del LCR no mostró alteraciones citoquímicas y los resultados del cultivo fueron negativos para bacterias, virus y hongos. Los resultados serológicos para enfermedades autoinmunes también fueron negativos. La RM evidenció afectación difusa supra e infratentorial, especialmente en los lóbulos frontales y el cuerpo calloso. También se observaron pequeñas zonas de realce y signos de hipertensión intracraneal (fig. 1 C-F).
En el diagnóstico diferencial clínico y radiológico se incluyeron varias enfermedades infecciosas, como la leucoencefalopatía multifocal progresiva y otras formas de encefalitis vírica; enfermedades autoinmunes como la encefalomielitis aguda diseminada, vasculitis o enfermedades del tejido conectivo; enfermedades metabólicas como algunas formas de leucodistrofia; la encefalopatía por radiación, y tumores cerebrales como la gliomatosis cerebri y el linfoma cerebral primario1,2.
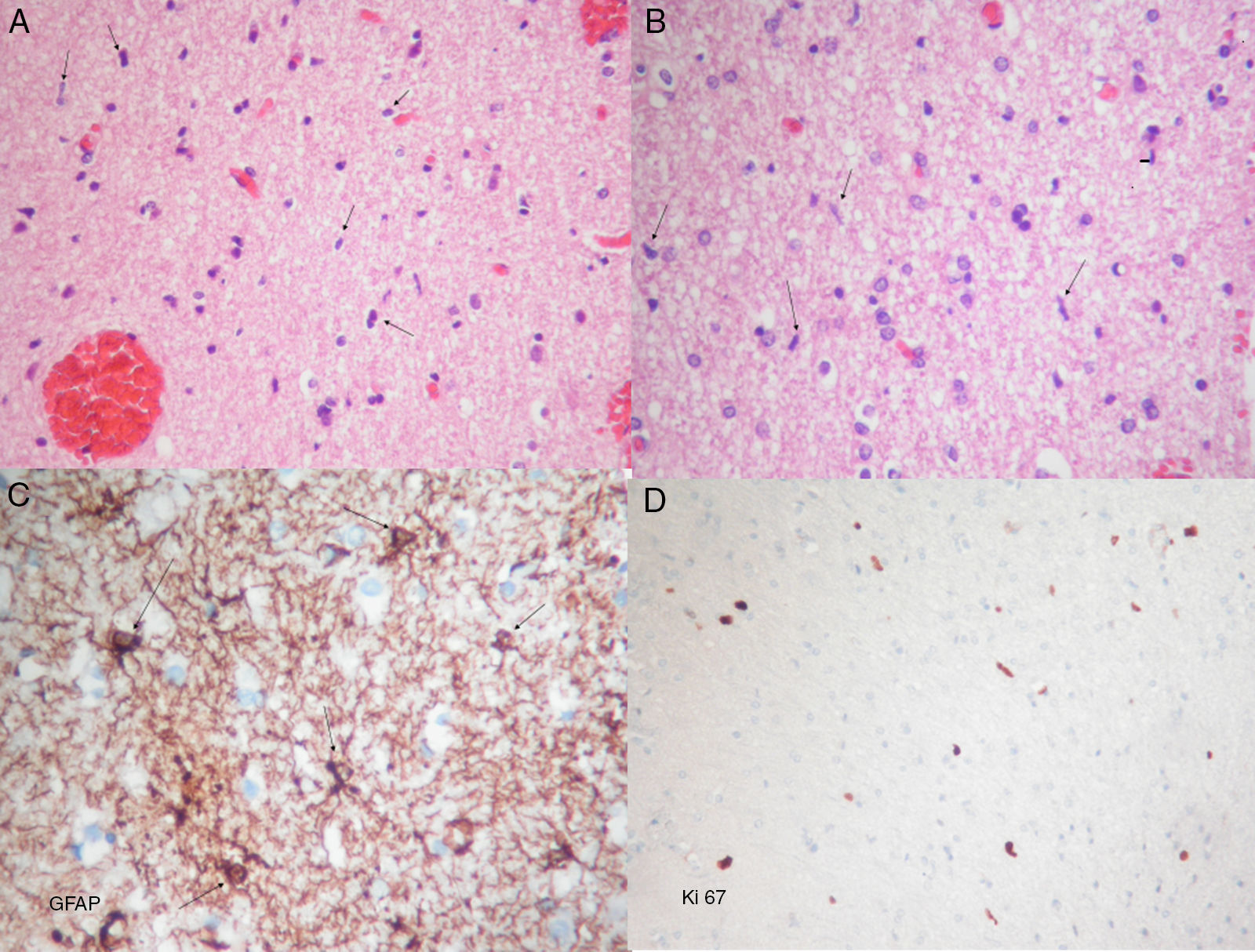
El análisis histopatológico mostró una proliferación astrocitaria difusa de baja densidad celular compatible con un tumor glial de bajo grado (fig. 2). Estos hallazgos, junto con los presentes en la RM, apuntaban hacia el diagnóstico de gliomatosis cerebri.
 Tinción con hematoxilina-eosina. Se observa una proliferación con celularidad moderada, caracterizada por la presencia de núcleos desnudos (flechas). B) Tinción con hematoxilina-eosina. La imagen muestra abundantes células de estirpe oligodendroglial. Elementos gliales con núcleos desnudos, fusiformes y moderadamente atípicos (flechas). C)Inmunohistoquímica para proteína gliofibrilar ácida (GFAP), que muestra elementos astrocíticos atípicos con extensiones cortas e irregulares (flechas). D) La tinción de Ki67 muestra una actividad proliferativa de aproximadamente el 2%. A, B y D: aumento original 10×; C: aumento original 40×.")
Imágenes de microscopía óptica.
A) Tinción con hematoxilina-eosina. Se observa una proliferación con celularidad moderada, caracterizada por la presencia de núcleos desnudos (flechas). B) Tinción con hematoxilina-eosina. La imagen muestra abundantes células de estirpe oligodendroglial. Elementos gliales con núcleos desnudos, fusiformes y moderadamente atípicos (flechas). C)Inmunohistoquímica para proteína gliofibrilar ácida (GFAP), que muestra elementos astrocíticos atípicos con extensiones cortas e irregulares (flechas). D) La tinción de Ki67 muestra una actividad proliferativa de aproximadamente el 2%. A, B y D: aumento original 10×; C: aumento original 40×.
A las 48 h de la cirugía, la paciente experimentó un aumento de la presión intracraneal refractario al tratamiento. De acuerdo con sus familiares, se decidió limitar el esfuerzo terapéutico. La paciente murió 6 días después de la cirugía.
La gliomatosis cerebri, que fue descrita por primera vez por Nevin en 19381, representa algo menos del 1% de los astrocitomas3,4. Es un trastorno neoplásico provocado por la infiltración de células gliales3,5,6 en el que al menos 3 lóbulos cerebrales se encuentran afectados1,3,6. Un rasgo característico de esta entidad es que mantiene la estructura macroscópica y la citoarquitectura del sistema nervioso central3,6.
Aunque se han descrito casos en niños7, suele presentarse en individuos de entre 40 y 50 años6. Su incidencia es ligeramente superior en los hombres3. La infiltración es predominantemente supratentorial, aunque con frecuencia se extiende a estructuras infratentoriales8-10. Suele afectar al cuerpo calloso, el tálamo y los ganglios basales6. También se ha descrito su extensión por todo el sistema nervioso central8.
La gliomatosis cerebri suele pasar desapercibida en fases iniciales, por lo que su diagnóstico se realiza por lo general en estadios avanzados7. Los primeros síntomas incluyen la aparición de focalidad neurológica o síntomas más inespecíficos, como cefalea, náuseas, vómitos, convulsiones, cambios de personalidad o deterioro cognitivo5.
Los hallazgos de neuroimagen en la gliomatosis cerebri son característicos pero, por lo general, inespecíficos9,10, lo que implica la necesidad de realizar un diagnóstico diferencial amplio. Normalmente las secuencias T2 y FLAIR de RM revelan áreas hiperintensas con una distribución asimétrica y/o heterogénea9,10. El cuerpo calloso suele estar afectado y se muestra engrosado9,10. Otra característica es la pérdida de la diferenciación entre la sustancia blanca y la gris9,10. En la gliomatosis cerebri tipo i no suele haber captación de contraste, mientras que en el tipo ii suelen apreciarse áreas realzadas que se relacionan con la transformación anaplásica9,10. La espectroscopia y las secuencias de volumen sanguíneo cerebral relativo pueden ayudar a reconocer el origen glial de estos tumores3. Estas secuencias también son útiles para escoger de una forma más precisa el área donde realizar la biopsia3.
Los rasgos histopatológicos de la gliomatosis cerebri suelen coincidir con los de un tumor glial de bajo grado5,6, aunque esta entidad puede evolucionar presentando rasgos propios de los tumores de alto grado5,6. El fenotipo celular más frecuente es astrocitario, aunque también puede ser oligodendrocítico u oligoastrocitario5. A pesar de su apariencia histológica, el comportamiento clínico de este tumor es similar al de un tumor de, al menos, grado iii, según la clasificación de la Organización Mundial de la Salud6.
El diagnóstico definitivo se alcanza tras examinar las características histopatológicas y de neuroimagen3,6. No obstante, la gran heterogeneidad de los hallazgos histológicos puede dificultar el diagnóstico3.
Dado que gliomatosis cerebri es una entidad infiltrativa y difusa, el papel de la cirugía se limita a la realización de biopsias con fines diagnósticos3,4. El tratamiento se basa fundamentalmente en la radioterapia11,12 y la quimioterapia13,14. Aun con esto, la mediana de supervivencia se sitúa entre los 14,5 y los 18 meses14,15. Los factores pronósticos de la gliomatosis cerebri son similares a los de otros gliomas3.
Nuestro caso representa un ejemplo de presentación clínica inicial devastadora de gliomatosis cerebri. Según nuestro conocimiento, esta es la primera vez que se describe en la literatura un caso de gliomatosis cerebri que se manifiesta con un síndrome de hipertensión intracraneal con coma resistente al tratamiento y con un fatídico desenlace.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses en lo que respecta a los materiales y métodos utilizados y los hallazgos reportados.

