




Paciente masculino de 26 años, con antecedentes de parto normal y pautas madurativas de desarrollo normales que alrededor de los 7 años inicia trastornos de inatención e hiperactividad motora. Se diagnostica trastorno por déficit atencional e hiperactividad (TDAH), siendo medicado con metilfenidato sin respuesta. Se le realizan estudios de IRM (imágenes de resonancia magnética) sin hallazgos patológicos. Evoluciona relativamente estable, pudiendo finalizar sus estudios secundarios. A los 17 años comienza con alucinaciones auditivas de tipo paranoide agregado a un trastorno de personalidad con agresividad. A esto se acompañó caídas de repetición con tendencia a la retropulsión, que mejoraron posteriormente. Se le diagnostica esquizofrenia paranoide. En 2010 desarrolla severo cuadro distónico en los 4 miembros de predominio izquierdo, con disartria y exacerbación de síntomas psiquiátricos que motiva la internación en la institución. Sus padres están sanos y son no consanguíneos; tiene dos hermanos sanos.
Examen neurológicoVigil conectado, comprende órdenes complejas a tres pasos, orientado parcialmente, nomina y repite con disartria severa. Risa inmotivada. El seguimiento ocular es fragmentado, con discreta limitación en la mirada vertical ascendente. El fondo de ojo es normal, con papilas de bordes netos, sin hallazgo de retinitis pigmentaria ni anillo de Kayser Fleischer; presenta una catarata incipiente bilateral. Sin asimetría facial sin déficit de pares bajos. Hipertonía rígida de los 4 miembros asociada a posturas distónicas, predominantemente del hemicuerpo izquierdo con el miembro superior en flexión, pronación y rotación interna. Hiperreflexia de los 4 miembros con predominio de los inferiores. Clonus agotable. Sensibilidad superficial y profunda conservada. Eumetría apendicular con temblor de fin de acción en la prueba índice-nariz. Marcha con severa retropulsión, discreto aumento de la base de sustentación y postura podálica en equinovaro.
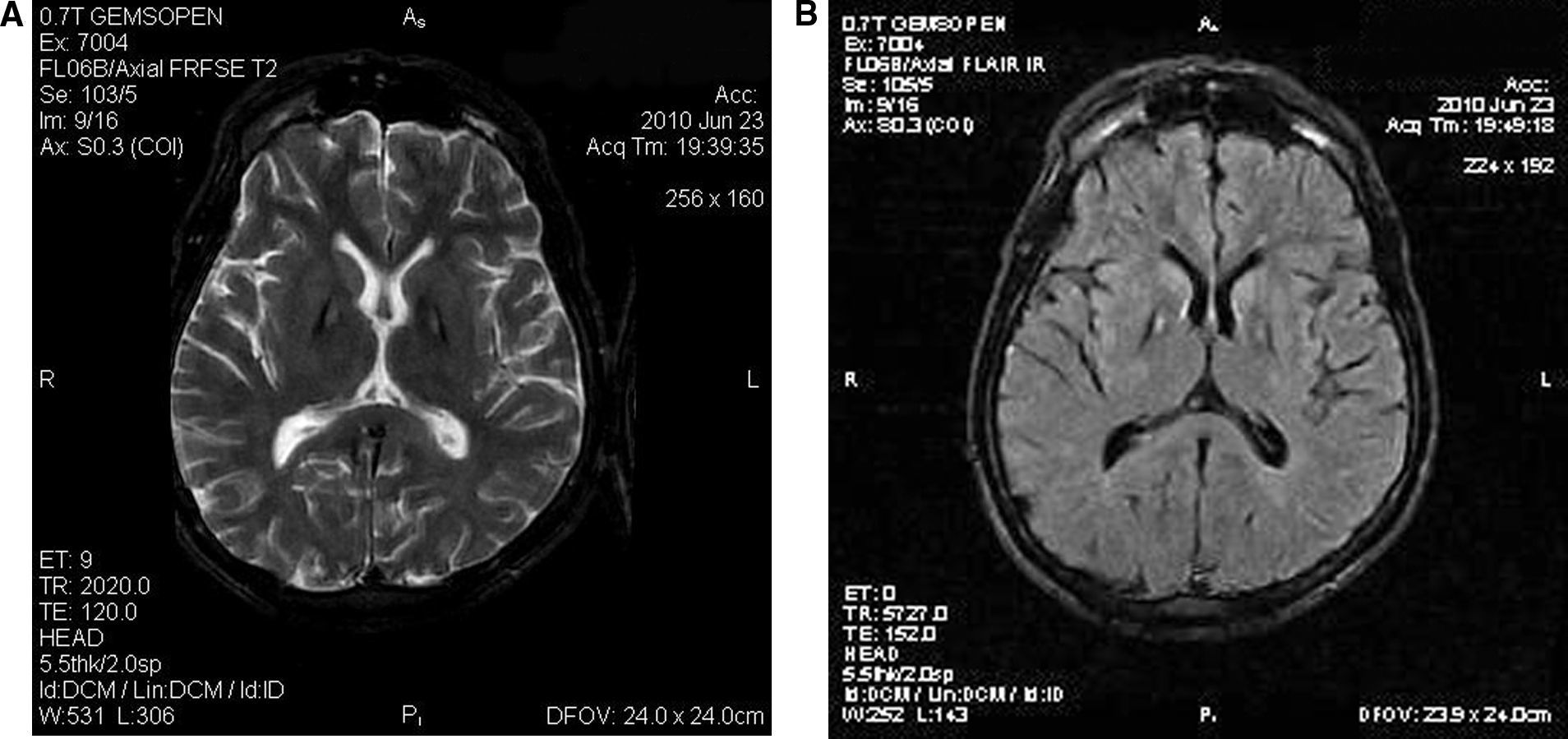
DiscusiónEl presente caso clínico describe un paciente joven que presenta como síntomas cardinales un severo cuadro rígido-acinético, distonía, disartria y trastornos psiquiátricos de forma fluctuante e inicio tardío, que unido a los hallazgos por IRM de encéfalo del «signo del ojo de tigre» (fig. 1) sugiere el diagnóstico de una forma atípica de neurodegeneración asociada a pantotenato quinasa (ex-enfermedad de Hallervorden-Spatz) (Pantothenate kinase associated neurodegeneration [PKAN en inglés]) [MIM234200]. PKAN se incluye dentro del complejo neurodegeneración asociada al depósito encefálico de hierro (Neurodegeneration with brain iron accumulation [NBIA en inglés]) que abarca un espectro de enfermedades que comparten entre sí la acumulación de hierro en el encéfalo, principalmente en los ganglios basales, y son causadas por defectos genéticos en el metabolismo de este metal. Su acumulación en el encéfalo genera un excesivo estrés oxidativo produciendo daño neuronal y muerte celular. Desde un punto de vista genético, las entidades definidas en este complejo son la neurodegeneración asociada al déficit de PKAN (por mutaciones en PKAN2), la neuroferritinopatía hereditaria (por mutaciones en FTL1), la distrofia neuroaxonal (por mutaciones en PLA2G6) y la aceruloplasminemia (por mutaciones CP). PKAN tiene dos formas de presentación, una típica o de inicio temprano y otra atípica o tardía. La forma típica se inicia antes de los 10 años, manifestándose principalmente como un síndrome distónico y trastornos de la marcha que llevan a la muerte de forma temprana, y se produce exclusivamente por mutaciones en el gen PKAN2. En la forma atípica predomina la disartria, el síndrome distónico y los trastornos de la personalidad; los síntomas se producen de forma más tardía y pueden presentar fluctuabilidad con periodos de agravamiento agudo vs. lapsos prolongados de estabilidad clínica1,2. Solo un porcentaje (alrededor de 35%) de las formas atípicas son producidas por PKAN21. Asumiendo por el relato familiar que el inicio de los síntomas sugestivos del diagnóstico de PKAN se produjeron entrando en la adolescencia, el curso evolutivo fluctuante, el hallazgo de imágenes normales durante su niñez y la ausencia de retinitis pigmentosa se podría afirmar que este caso corresponde a una forma atípica o tardía. No podemos afirmar con certeza si el trastorno atencional-conductual que presentó desde niño correspondería a una condición independiente o a un síntoma incipiente de la enfermedad.
 y FLAIR (B) respectivamente. En secuencia T2 y FLAIR se observan lesiones simétricas bipalidales, hiperintensas con un halo hipointenso circundante (signo del «ojo de tigre»). El resto del parénquima encefálico es de características normales.")
IRM de encéfalo; imágenes axiales ponderadas en T2 (A) y FLAIR (B) respectivamente. En secuencia T2 y FLAIR se observan lesiones simétricas bipalidales, hiperintensas con un halo hipointenso circundante (signo del «ojo de tigre»). El resto del parénquima encefálico es de características normales.
Desde el punto de vista imagenológico el «signo del ojo de tigre» es un signo radiológico visualizado fácilmente por IRM que denota la acumulación del hierro de forma simétrica en la región medial de ambos globos pálidos, produciendo una señal hipointensa asociada a un halo hiperintenso en la secuencia T2. La presencia aislada de este signo, acompañado o no de hipointensidad de la sustancia nigra, sería patognomónico de los casos de PKAN causados por mutaciones en PKAN21,3,4. Se ha descrito este signo en aislados casos de neuroferritinopatía, pero los mismos estaban asociados a hiperintensidad en otras regiones del encéfalo como el córtex cerebral, el putamen, el núcleo dentado e hipointensidad en los tálamos5.
Excluyendo las imágenes, y desde el punto de vista clínico-semiológico, existen numerosas entidades que deberían ser consideradas como diagnóstico diferencial de un síndrome rígido acinético asociado a fenómenos distónicos y cambios de la personalidad de inicio en la juventud. Los mismos incluyen la enfermedad de Parkinson juvenil, la enfermedad de Wilson, la enfermedad de Huntington (variante rígido acinética de Westhpal), la neuroacantocitosis, la enfermedad de Fahr de inicio juvenil y la forma tardía de la enfermedad de Tay Sachs, entre otras.
El traslado repentino a otra institución y la negativa de los padres no permitió realizar el estudio genético del paciente, además de poder realizar otros estudios complementarios para descartar los diagnósticos diferenciales mencionados. En resumen, el presente reporte describe a un paciente que tanto por las imágenes como por la clínica sugiere que es portador de una PKAN atípica o tardía, caracterizado por un síndrome rígido acinético, distonía y disartria más la presencia aislada del «signo del ojo de tigre» en la IRM del encéfalo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
