




Dentro de las enfermedades mitocondriales la oftalmoplejía externa crónica progresiva (CPEO) constituye una entidad clínica definida. Sus patrones de herencia son complejos, describiéndose formas esporádicas, formas con transmisión materna y formas mendelianas. Tanto las formas mendelianas tipo dominante (adCPEO) o recesivas (arCPEO) están producidas por mutaciones en distintos genes nucleares, entre ellos POLG1 y POLG2, C10orf2 (PEO1), ANT1 y OPA1. C10orf2 codifica para la enzima helicasa «Twinkle» y junto a POLG γ participa en la replicación del ADN mitocondrial (ADNmt). Fallos en estos mecanismos conducen a inestabilidad del mismo y a la producción de múltiples deleciones.
ObjetivoSe presenta un caso de adCPEO en una familia argentina y el hallazgo una probable mutación en PEO1, no descripta previamente en la literatura mundial.
Caso clínicoPaciente de 49 años que comienza a los 14 con ptosis palpebral, desarrollando a los 20 oftalmoplejía sin diplopía. En el último tiempo agregó debilidad muscular proximal y distal asociada a disfagia e hipofonía. La historia familiar cuenta con similares síntomas en la rama paterna (abuela, tía, padre, hermano).
ConclusiónMutaciones en C10Orf2 (PEO1) deben ser consideradas como causa frecuente de las formas de CPEO dominante, así como aquellas producidas en otros genes conocidos como POLG, POLG2, ANT1 y eventualmente OPA1. En las formas clínicas asociadas PEO1 habitualmente no existe compromiso multisistémico. En el contexto clínico adecuado, el estudio genético estaría indicado como primer paso en el algoritmo de estudio de estos pacientes, evitando intervenciones invasivas como la biopsia muscular. La caracterización genética de estas enfermedades permitiría ofrecer un diagnóstico y consejo genético adecuado, así como predecir relativamente la evolución de la enfermedad.
Among mitochondrial disorders, chronic progressive external opthalmoplegia (CPEO) conforms a define entity. Inheritance patterns are complex, describing sporadic cases, maternally inherited and mendelian forms. Both, autosomal dominant (adCPEO) and recesive (arCPEO) forms are due to mutations in different nuclear genes like POLG1, POLG2, C10orf2 (PEO1), ANT1 and OPA1. C10orf2 codifies for the helicase «Twinkle» and along with POLG γ participates in the replication of mitochondrial DNA (mtDNA). Defects on these mechanisms lead to instability of mtDNA and consequently, the production of multiple deletions.
ObjetiveWe present a case of a probable novel mutation in PEO1 in an Argentinean family with adCPEO.
Case report49 year old woman with history of ptosis since 14, developing progressive opthalmoplegia at 20 without diplopia. Recently she added generalized muscle weakness associated with dysphagia and voice changes. She has a family history with similar symptoms in the paternal line.
ConclusionMutations in C10Orf2 (PEO1) should be considered a frequent cause of adCPEO as well as POLG, POLG2, ANT1 and eventual OPA1. CPEO associated with mutations in PEO1 cause a mild phenotype, usually without multisystem involvement. In the appropriate clinical context, genetic studies can avoid invasive interventions like a muscle biopsy and give an appropriate diagnosis. Genetic characterization of these disorders can offer a proper genetic counseling along with a relative prognosis of the disease.
La oftalmoplejía crónica progresiva (CPEO) es un síndrome clínico definido dentro del espectro de las enfermedades mitocondriales y constituye un modelo paradigmático en la compresión de los distintos patrones de herencia que pueden seguir estas complejas enfermedades. Existen formas esporádicas producidas por mutaciones espontáneas (grandes reordenamientos) originadas en estadios tempranos del desarrollo embriogénico u oogénico1,2, formas trasmitidas por vía materna habitualmente producidas por mutaciones puntuales en los ARN de transferencia (ARNt)3–5 y formas mendelianas6–8. Estas últimas son producidas por mutaciones en genes nucleares, pudiendo seguir un patrón de herencia autosómico dominante (adCPEO) o recesivo (arCPEO). Los genes involucrados son aquellos que intervienen en la llamada «comunicación intergenómica» entre el ADN nuclear (ADNn) y el mitocondrial (ADNmt); POLG1 y POLG2, codificando para la enzima POLGγ, SLC25A4 codificando para la enzima ANT1 y C10Orf2 (PEO1), para la enzima Twinkle. En conjunto, conforman el aparato replicativo del ADNmt9. Fallos en las proteínas codificadas por dichos genes llevan a dificultades en la replicación y estabilidad mitocondrial, y por consiguiente, a la acumulación de mutaciones en el ADNmt replicado produciendo un patrón de múltiples deleciones visibles por técnicas moleculares10. Además, otros genes como OPA1, que participa en la organización miotubular y fusión mitocondrial, han sido vinculados con formas de CPEO y atrofia óptica11,12, así como el gen RRM2B.
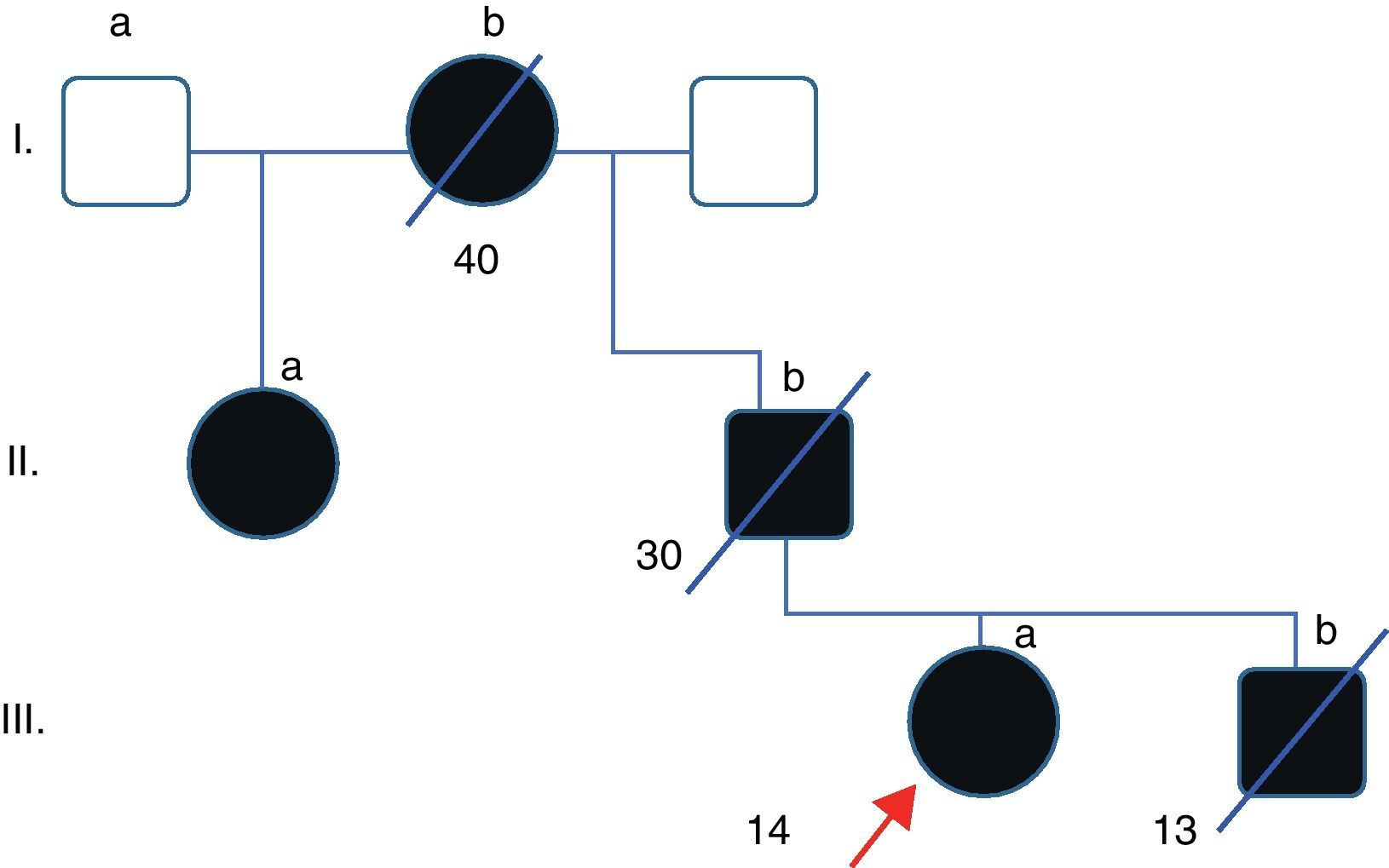
Desde el punto de vista clínico la CPEO se caracteriza por la instalación progresiva de ptosis palpebral y oftalmoparesia bilateral, pudiendo ser asimétrica y de instalación más frecuentemente en la tercera a cuarta década de la vida13, aunque existen formas de presentación temprana y formas de inicio más allá de los 60 años. Pueden presentar distinto grado de debilidad muscular proximal y orofaríngea con consiguiente disfagia. En las formas esporádicas de CPEO causadas por grandes reordenamientos es menos frecuente el compromiso cardíaco típico del síndrome de Kearns Sayre14. Las formas mendelianas de CPEO causadas por mutaciones en POLG γ habitualmente se manifiestan como enfermedades multisistémicas, donde la combinación de síntomas puede incluir parkinsonismo atípico, distonía, neuropatía sensitivomotora axonal o desmielinizante, ganglionopatía, entre otros (ver www.mitomap.org/POLG database). Presentamos el primer caso argentino de adCPEO y el hallazgo de una probable mutación en PEO1, no descrita previamente en la literatura mundial.
Caso clínicoPaciente de 49 años que acude a la consulta relatando ptosis palpebral de lenta progresión desde los 14 años, desarrollando posteriormente oftalmoplejía bilateral y simétrica sin diplopía. Dichos síntomas fueron progresando hasta agregar en su adultez dificultades para elevar los hombros y sostener objetos pesados, cansancio generalizado así como disfagia e hipofonía. En estos últimos años fue diagnosticada de depresión ansiosa. No refiere trastornos en la audición ni disminución de la agudeza visual. Presenta historia familiar con similares síntomas: abuela paterna con oftalmoplejía y ptosis de inicio alrededor de los 40 años, padre con oftalmoplejía de inicio a los 30 años, tía (media hermana paterna) con oftalmoplejía de inicio adulto. El hermano de la paciente está afectado con inicio de similares síntomas desde los 13 años (fig. 1). Todos los familiares del caso índice han fallecido por diversas causas. En el examen físico se constata una paciente lúcida (MMSE 29/30), con oftalmoplejía completa en todos los planos de la mirada y reflejos pupilares conservados, ptosis bilateral (fig. 2). Presenta debilidad lingual y voz hipofónica y relata trastornos deglutorios para sólidos. Debilidad muscular predominantemente en músculos proximales (flexores del cuello y cinturas) así como dificultad para caminar sobre los talones, con hiporreflexia generalizada. Moderada atrofia muscular generalizada; sensibilidad y taxia conservada. Se realizó un análisis de laboratorio control en la paciente, descartándose diabetes u otros trastornos metabólicos/endocrinológicos. El examen auditivo y cardiológico y la RMN de encéfalo no mostraron alteraciones.
.")
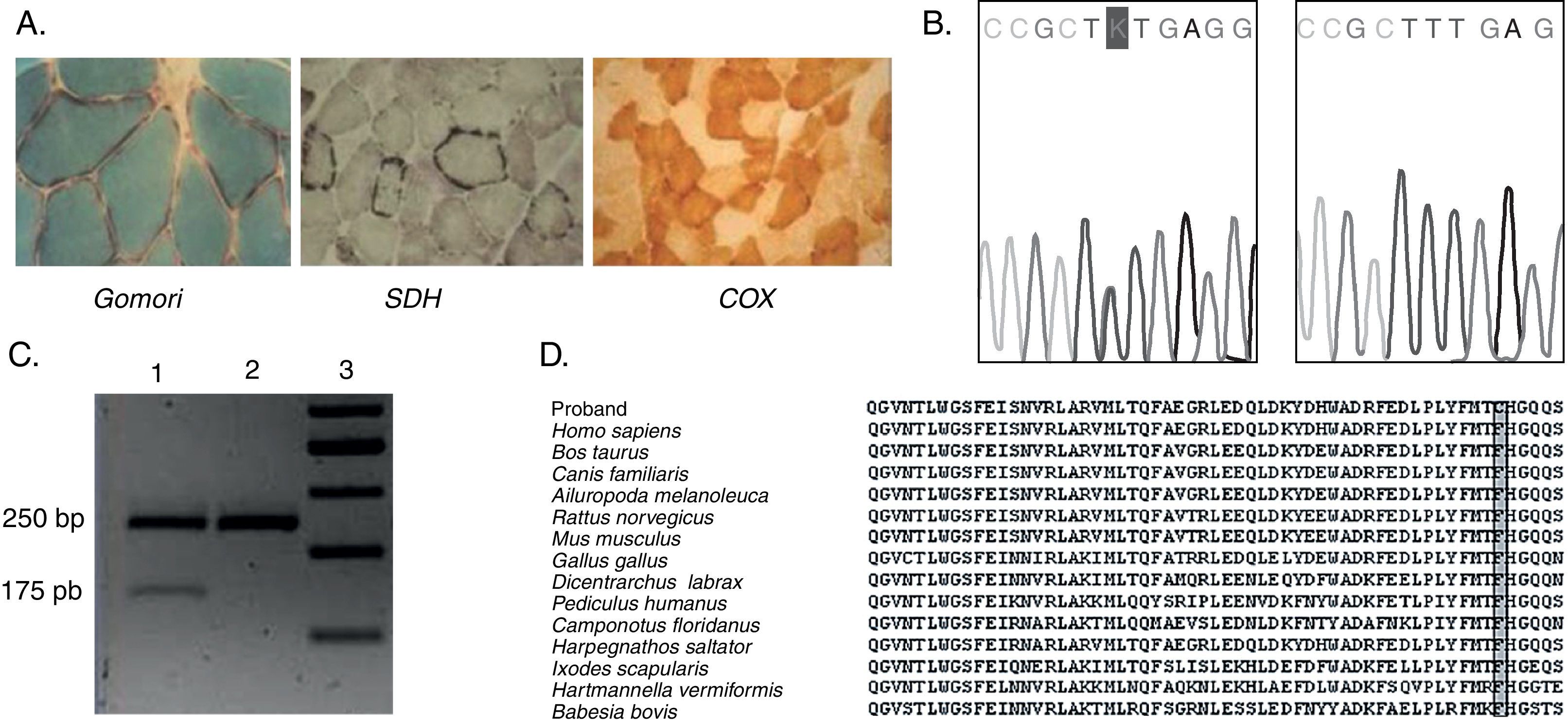
![A. Cortes histológicos de músculo del caso III.b (fig. 1). Las distintas tinciones muestran la presencia de fibras rojas rasgadas (FRR) con la técnica de Gomori, incremento subsarcolemal de mitocondrias anormales mediante la técnica de succinato dehidrogenasa (SDH) y la presencia de fibras citocromo oxidasa negativas (en blanco) con la tinción para COX. B. Electroferograma mostrando el cambio heterocigotico c.1618T>G (rectángulo) en el gen PEO1, en ADN extraído de sangre del paciente (izquierda) (paciente IIIa [fig. 1]) vs. Secuencia wild type(derecha). C. Análisis por técnica de PCR-RFLP de un fragmento de ADN que incluye la mutación mencionada. El cambio es reconocido por la enzima de restricción MspA1I en presencia de la mutación; carril 1, probando; carril 2, control; carril 3, marcador de peso molecular de ADN. D. Conservación evolutiva del residuo fenilalanina en la posición 478 de la proteína «Twinkle» (rectángulo gris).](https://static.elsevier.es/multimedia/18530028/0000000400000003/v1_201305151237/S1853002812000249/v1_201305151237/es/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcnxJVYM2BEAEuZ5XZLhi9iaMnPzhM4IXd3zNtr3zSujuuxL5uy3FKNlxjodSd5rYNzJw2QBNngdobesGMNjB5yvEBVk2e8AQmkCcfmtyhY+Uyj52eQHq/pj+6F78J1kwCyAWko0LSNqAt+Huzwo98tDnyKkfJZB4TRTCBoY9nWetuHU/LQIVkbNvJgdALC8ZatJjGxouVPDtR35yI/S7OchVxpCT3gTlfepEULp8Xnbq1rduS506uXVjJ5RbEi9Og= "A. Cortes histológicos de músculo del caso III.b (fig. 1). Las distintas tinciones muestran la presencia de fibras rojas rasgadas (FRR) con la técnica de Gomori, incremento subsarcolemal de mitocondrias anormales mediante la técnica de succinato dehidrogenasa (SDH) y la presencia de fibras citocromo oxidasa negativas (en blanco) con la tinción para COX. B. Electroferograma mostrando el cambio heterocigotico c.1618T>G (rectángulo) en el gen PEO1, en ADN extraído de sangre del paciente (izquierda) (paciente IIIa [fig. 1]) vs. Secuencia wild type(derecha). C. Análisis por técnica de PCR-RFLP de un fragmento de ADN que incluye la mutación mencionada. El cambio es reconocido por la enzima de restricción MspA1I en presencia de la mutación; carril 1, probando; carril 2, control; carril 3, marcador de peso molecular de ADN. D. Conservación evolutiva del residuo fenilalanina en la posición 478 de la proteína «Twinkle» (rectángulo gris).")
A. Cortes histológicos de músculo del caso III.b (fig. 1). Las distintas tinciones muestran la presencia de fibras rojas rasgadas (FRR) con la técnica de Gomori, incremento subsarcolemal de mitocondrias anormales mediante la técnica de succinato dehidrogenasa (SDH) y la presencia de fibras citocromo oxidasa negativas (en blanco) con la tinción para COX. B. Electroferograma mostrando el cambio heterocigotico c.1618T>G (rectángulo) en el gen PEO1, en ADN extraído de sangre del paciente (izquierda) (paciente IIIa [fig. 1]) vs. Secuencia wild type(derecha). C. Análisis por técnica de PCR-RFLP de un fragmento de ADN que incluye la mutación mencionada. El cambio es reconocido por la enzima de restricción MspA1I en presencia de la mutación; carril 1, probando; carril 2, control; carril 3, marcador de peso molecular de ADN. D. Conservación evolutiva del residuo fenilalanina en la posición 478 de la proteína «Twinkle» (rectángulo gris).
La biopsia muscular practicada, numerosos años atrás en el hermano (fig. 1 [caso III.b]) de la paciente, mostró la presencia de abundantes fibras ragged red y COX deficitarias, así como incremento subsarcolemal en las tinciones SDH, sugiriendo el diagnóstico histológico de una patología mitocondrial (fig. 2A). Años después del diagnóstico histológico la paciente es citada para completar estudios moleculares. Se procede al análisis por secuenciación del ADN, extraído de la sangre de la paciente previo a la firma del consentimiento informado, del gen POLG1, descartándose alteraciones. Posteriormente se decide secuenciación de genes ANT1 y C10orf2. Este último presenta un cambio nucleotídico en la posición c.1618T>G (exón 2) prediciendo la sustitución aminoacídica p.F478C que genera el paso de un aminoácido apolar a uno polar (fig. 2B). Este cambio no ha sido reportado en la literatura mundial ni como un polimorfismo ni como una mutación conocida, pudiendo corresponder a una mutación missense en heterocigosis no descrita previamente. Lamentablemente no pudo realizarse el análisis segregacional, ya que el resto de los familiares afectados se encontraban fallecidos en el momento de la primera entrevista.
DiscusiónSe presentó un caso de adCPEO en el cual se realizó estudio molecular con el hallazgo del cambio nucleotídico en la posición c.1618T>G (p.F478C) del gen PEO1. A pesar de no poder realizarse el análisis segregacional existen argumentos para considerar que el mismo es la causa del fenotipo descripto: a) el cambio aminoacídico es marcado (apolar a polar); b) dicho cambio no ha sido reportado como un polimorfismo neutral en las bases consultadas, ni tampoco en los 200 controles sanos en los que se ha realizado; c) tomando como referencia que todas las mutaciones descritas previamente en Co10Orf2 (PEO1) se encuentran en los exones 1 y 2, el cambio se encuentra presente en el dominio helicasa del exón 2, en contigüidad a la mutación p.E479K; d) la base se encuentra altamente preservada en las distintas especies, alejando la posibilidad de un polimorfismo; y e) el cambio de base fue confirmado mediante la técnica de RFLP.
Las formas mendelianas de CPEO están producidas por mutaciones en distintos genes nucleares, entre ellos POLG1-2, C10orf2 (PEO1), ANT1 y OPA1. PEO1 codifica para la enzima helicasa «Twinkle» y junto a POLG γ participa en la replicación del ADNmt. Fallos en estos mecanismos conducen a inestabilidad del ADNmt y a la producción de deleciones en el mismo. Clínicamente la oftalmoparesia constituye el signo-síntoma cardinal en la patología mitocondrial, aunque la misma puede acompañarse de manifestaciones sistémicas como neuropatía, debilidad muscular, depresión, cataratas, cardiomiopatía y rara vez parkinsonismo15. Un dato particular en este caso es que parecería existir un fenómeno anticipatorio transgeneracional, tal como se describe en las patologías causadas por expansiones nucleotídicas. Esta observación ha sido reportada previamente en mutaciones causadas en POLG116, aunque no constituiría un rasgo frecuente. Aislados reportes de ataxia recesiva de inicio infantil, así como síndromes miohepatoencefálicos (síndrome de Alpers) por mutaciones en C10orf2 han sido descritos17,18. A diferencia de POLG, donde se han descrito variadas formas clínicas, incluso sin oftalmoplejía asociada (ver. www.mitomap.org/POLG database), las mutaciones en C10orf2 tenderían a producir un fenotipo más uniforme, con menos compromiso multisistémico (formas de adCPEO «puras») y a diferencia del caso comentado, su inicio se produce en etapas mas tardías de la vida.
En conclusión, se describe un caso de adCPEO en una familia argentina, con el hallazgo de un cambio nucleotídico que correspondería a mutación no descrita previamente en la literatura mundial, asociado a un aparente fenómeno anticipatorio transgeneracional. En el contexto clínico adecuado el estudio genético estaría indicado como primer paso en el algoritmo de estudio de pacientes con adCPEO, ya que la ausencia de signos morfológicos característicos en la biopsia muscular no descarta el diagnóstico. La caracterización genética de estas enfermedades permite acceder al diagnóstico en aquellos casos donde la biopsia no es informativa, además de ofrecer un consejo genético.
FinanciaciónEste trabajo recibió el apoyo del Ministerio de Ciencia e Innovación del Gobierno de España, ISCIII, PI09-1359. El Dr. Martin recibe financiación del programa «Intensificación de la actividad investigadora» de la Agencia ISCIII-Laín Entralgo CM, Madrid.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores agradecen a la paciente su colaboración desinteresada. El Dr. Berardo agradece a Cecilia Van Cauwelaert la asistencia técnica en la composición de las figuras. Los Dres. Martin y Rivera agradecen a Sara Jiménez su asistencia técnica.
